Comparison of all ChIP Targets
Stephen Pederson and Richard Iggo
22 June, 2022
Last updated: 2022-06-22
Checks: 6 1
Knit directory:
apocrine_signature_mdamb453/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220427) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 4adda39. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/bigwig/
Untracked files:
Untracked: analysis/comparisonRI.Rmd
Unstaged changes:
Modified: analysis/comparison.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/comparison.Rmd) and HTML
(docs/comparison.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 4adda39 | Steve Pederson | 2022-06-08 | Corrected promoter/enhancer count discrepancy |
| html | 4adda39 | Steve Pederson | 2022-06-08 | Corrected promoter/enhancer count discrepancy |
| Rmd | 1f959c3 | Steve Pederson | 2022-06-08 | Finished checking motif distances |
| html | 1f959c3 | Steve Pederson | 2022-06-08 | Finished checking motif distances |
| Rmd | b882cb8 | Steve Pederson | 2022-06-07 | Ranges are no longer centred |
| Rmd | e1c43dd | Steve Pederson | 2022-06-06 | Tidied genomic plots |
| html | e1c43dd | Steve Pederson | 2022-06-06 | Tidied genomic plots |
| Rmd | 480c17a | Steve Pederson | 2022-06-06 | Corrected typo |
| html | 480c17a | Steve Pederson | 2022-06-06 | Corrected typo |
| Rmd | e1088ac | Steve Pederson | 2022-06-03 | revised comparison |
| html | e1088ac | Steve Pederson | 2022-06-03 | revised comparison |
| Rmd | cf12cba | Steve Pederson | 2022-06-01 | Moved AR to the correct section |
| html | cf12cba | Steve Pederson | 2022-06-01 | Moved AR to the correct section |
| Rmd | a6276af | Steve Pederson | 2022-06-01 | Changed HiC plot names to HiChIP |
| html | a6276af | Steve Pederson | 2022-06-01 | Changed HiC plot names to HiChIP |
| Rmd | 8836090 | Steve Pederson | 2022-05-26 | Added motif centrality |
| html | 8836090 | Steve Pederson | 2022-05-26 | Added motif centrality |
| html | 65e26b8 | Steve Pederson | 2022-05-20 | Build site. |
| Rmd | 3a37987 | Steve Pederson | 2022-05-20 | Finished first draft without motifs |
| Rmd | 994ec9a | Steve Pederson | 2022-05-20 | Added enrichment testing |
| html | 994ec9a | Steve Pederson | 2022-05-20 | Added enrichment testing |
| Rmd | 5597186 | Steve Pederson | 2022-05-19 | Added MYC plot |
| Rmd | 1ba686b | Steve Pederson | 2022-05-19 | EOD Wed |
| Rmd | e742af3 | Steve Pederson | 2022-05-16 | Started exploring mapped genes |
| html | e742af3 | Steve Pederson | 2022-05-16 | Started exploring mapped genes |
| Rmd | 792bb98 | Steve Pederson | 2022-05-13 | Added dist to TSS |
| html | 792bb98 | Steve Pederson | 2022-05-13 | Added dist to TSS |
| Rmd | e9956d4 | Steve Pederson | 2022-05-13 | Added 95% CIs for targets |
| html | e9956d4 | Steve Pederson | 2022-05-13 | Added 95% CIs for targets |
| Rmd | 1582607 | Steve Pederson | 2022-05-12 | Started looking at H3K27ac |
| Rmd | 8217e8a | Steve Pederson | 2022-05-10 | Started explorations |
| html | 8217e8a | Steve Pederson | 2022-05-10 | Started explorations |
Introduction
library(tidyverse)
library(magrittr)
library(extraChIPs)
library(plyranges)
library(pander)
library(scales)
library(reactable)
library(htmltools)
library(UpSetR)
library(rtracklayer)
library(GenomicInteractions)
library(multcomp)
library(glue)
library(nnet)
library(effects)
library(Gviz)
library(readxl)
library(corrplot)
library(goseq)
library(cowplot)
library(scico)
library(BSgenome.Hsapiens.UCSC.hg19)
theme_set(theme_bw())bar_chart <- function(label, width = "100%", height = "16px", fill = "#00bfc4", background = NULL) {
bar <- div(style = list(background = fill, width = width, height = height))
chart <- div(style = list(flexGrow = 1, marginLeft = "8px", background = background), bar)
div(style = list(display = "flex", alignItems = "center"), label, chart)
}
bar_style <- function(width = 1, fill = "#e6e6e6", height = "75%", align = c("left", "right"), color = NULL) {
align <- match.arg(align)
if (align == "left") {
position <- paste0(width * 100, "%")
image <- sprintf("linear-gradient(90deg, %1$s %2$s, transparent %2$s)", fill, position)
} else {
position <- paste0(100 - width * 100, "%")
image <- sprintf("linear-gradient(90deg, transparent %1$s, %2$s %1$s)", position, fill)
}
list(
backgroundImage = image,
backgroundSize = paste("100%", height),
backgroundRepeat = "no-repeat",
backgroundPosition = "center",
color = color
)
}dht_peaks <- here::here("data", "peaks") %>%
list.files(recursive = TRUE, pattern = "oracle", full.names = TRUE) %>%
sapply(read_rds, simplify = FALSE) %>%
lapply(function(x) x[["DHT"]]) %>%
lapply(setNames, nm = c()) %>%
setNames(str_extract_all(names(.), "AR|FOXA1|GATA3|TFAP2B"))
targets <- names(dht_peaks)
sq <- seqinfo(dht_peaks[[1]])
dht_peaks %>%
lapply(mutate, w = width) %>%
lapply(as_tibble) %>%
bind_rows(.id = "target") %>%
ggplot(aes(as.factor(n_reps), w, fill = target)) +
geom_violin(draw_quantiles = 0.5) +
scale_y_log10() +
labs(x = "Number of Replicates", y = "Peak Width", fill = "Target")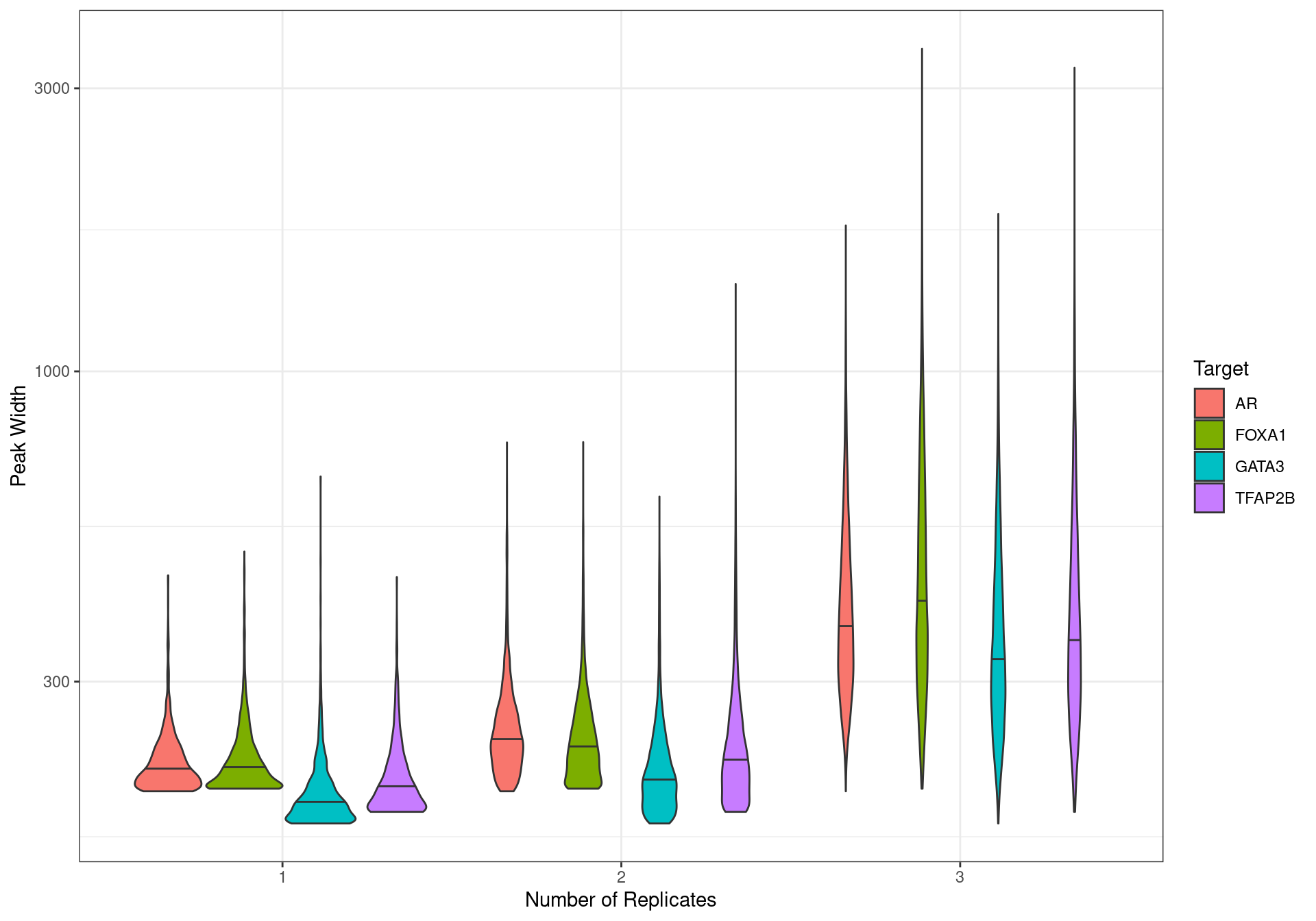
Peak width distribution based on the number of replicates a peak was detected in.
| Version | Author | Date |
|---|---|---|
| 1f959c3 | Steve Pederson | 2022-06-08 |
Given that narrow peaks were primarily associated with those detected in a single replicate, the decision was made to only retain peaks detected in \(\geq2\) replicates for each target. This would remove the following peak numbers from each target
- AR: 1,326 (6.8%)
- FOXA1: 1,872 (2.0%)
- GATA3: 1,245 (4.2%)
- TFAP2B: 691 (3.3%)
cp <- htmltools::tags$em(
"
Summary of all oracle peaks from DHT-treated samples.
FOXA1 clearly showed the most binding activity.
Discarded peaks are those only detected in one replicate.
"
)
tbl <- dht_peaks %>%
lapply(
function(x) {
tibble(
n = sum(x$n_reps > 1),
w = median(width(x)),
kb = sum(width(x)) / 1e3,
discarded = sum(x$n_reps == 1)
)
}
) %>%
lapply(list) %>%
as_tibble() %>%
pivot_longer(cols = everything(), names_to = "target") %>%
unnest(everything()) %>%
reactable(
filterable = FALSE, searchable = FALSE,
columns = list(
target = colDef(name = "ChIP Target"),
n = colDef(name = "Total Peaks"),
w = colDef(name = "Median Width"),
kb = colDef(name = "Total Width (kb)", format = colFormat(digits = 1)),
discarded = colDef(name = "Discarded Peaks")
),
defaultColDef = colDef(
format = colFormat(separators = TRUE, digits = 0)
)
)
div(class = "table",
div(class = "table-header",
div(class = "caption", cp),
tbl
)
)all_gr <- here::here("data", "annotations", "all_gr.rds") %>%
read_rds()
tss <- resize(all_gr$transcript, width = 1)
features <- here::here("data", "h3k27ac") %>%
list.files(full.names = TRUE, pattern = "bed$") %>%
sapply(import.bed, seqinfo = sq) %>%
lapply(granges) %>%
setNames(basename(names(.))) %>%
setNames(str_remove_all(names(.), "s.bed")) %>%
GRangesList() %>%
unlist() %>%
names_to_column("feature") %>%
sort()
dht_consensus <- dht_peaks %>%
lapply(filter, n_reps > 1) %>%
lapply(mutate, w = width) %>%
lapply(
function(x) {
x <- shift(x, x$peak)
x <- resize(x, width = 1, fix = 'start')
resize(x, width = round(0.9*x$w, 0), fix = 'center')
}
) %>%
lapply(granges) %>%
GRangesList() %>%
unlist() %>%
reduce()%>%
mutate(
AR = overlapsAny(., dht_peaks$AR),
FOXA1 = overlapsAny(., dht_peaks$FOXA1),
GATA3 = overlapsAny(., dht_peaks$GATA3),
TFAP2B = overlapsAny(., dht_peaks$TFAP2B),
H3K27ac = propOverlap(., features) > 0.5,
promoter = bestOverlap(., features, var = "feature") == "promoter" & H3K27ac,
enhancer = bestOverlap(., features, var = "feature") == "enhancer" & H3K27ac,
dist_to_tss = mcols(distanceToNearest(., tss, ignore.strand = TRUE))$distance
)Consensus peaks were then formed by:
- Centering each target’s peak-set at the peak summit
- Resizing peaks to 90% of the initial width
- Merging any overlapping peaks from multiple targets
Target overlap was defined as the merged peak having any overlap with the set of original peaks from each target. H3K27ac overlap was considered to be true if >50% of the final peak overlapped an H3K27ac-marked region.
Relationship Between Binding Regions
Oracle peaks from the DHT-treated samples in each ChIP target were obtained previously using the GRAVI workflow. AR and GATA3 peaks were derived from the same samples/passages, whilst FOXA1 and TFAP2B ChIP-Seq experiments were performed separately.
A set of target-agnostic set of binding regions was then defined as the union of all DHT-treat peaks across all targets.
dht_consensus %>%
as_tibble() %>%
pivot_longer(cols = all_of(targets), names_to = "target", values_to = "bound") %>%
dplyr::filter(bound) %>%
split(.$target) %>%
lapply(pull, "range") %>%
fromList() %>%
upset(
sets = rev(targets), keep.order = TRUE,
order.by = "freq",
set_size.show = TRUE, set_size.scale_max = nrow(.)
)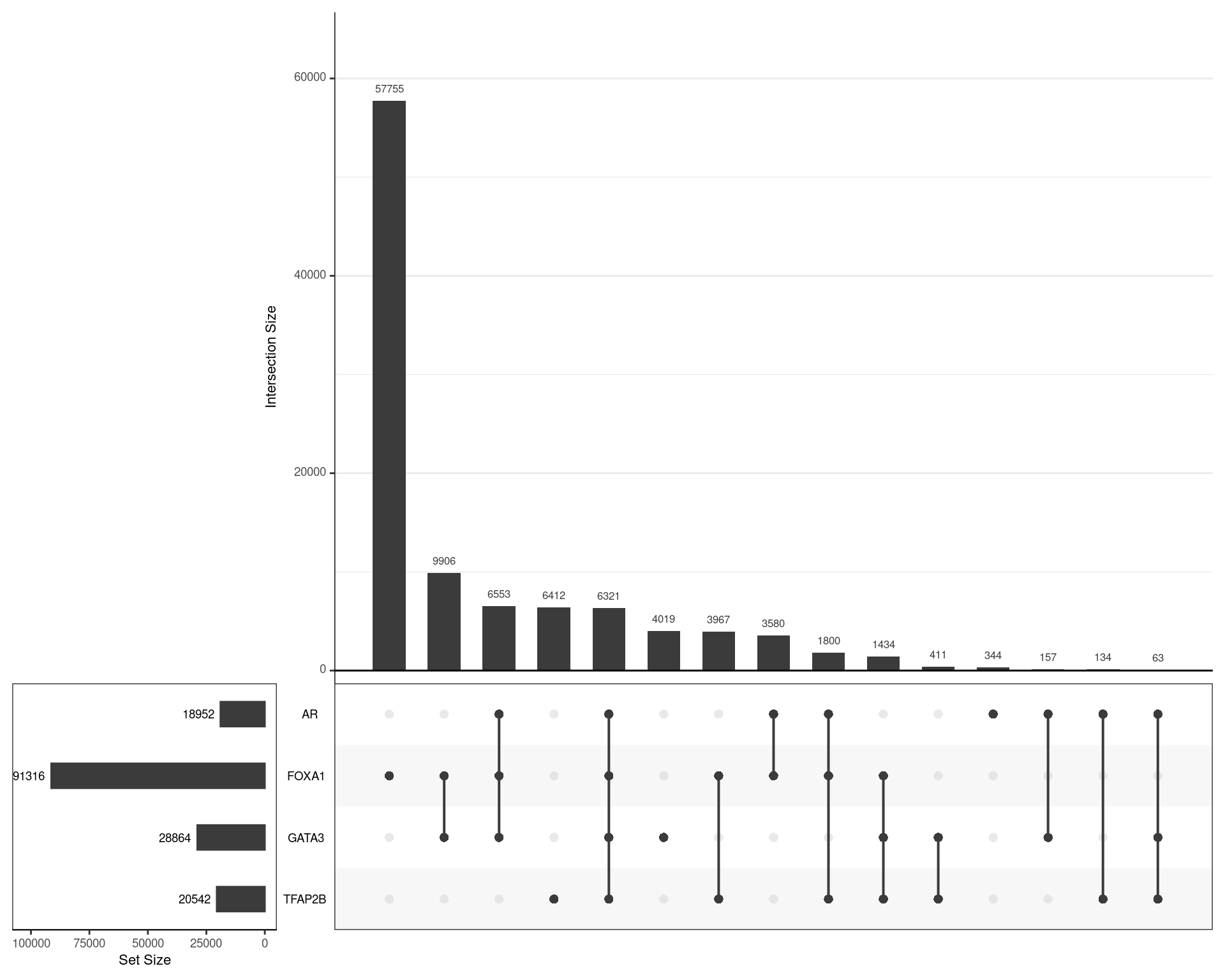
Using the union of all binding regions, those which overlapped an oracle peak from each ChIP target are shown
Peak Width
df <- dht_consensus %>%
mutate(w = width) %>%
as_tibble() %>%
pivot_longer(
cols = all_of(targets),
names_to = "targets", values_to = "detected"
) %>%
dplyr::filter(detected) %>%
group_by(range, H3K27ac, w) %>%
summarise(
targets = paste(sort(targets), collapse = " "),
.groups = "drop"
)
df %>%
mutate(n_targets = str_count(targets, " ") + 1) %>%
arrange(n_targets, targets) %>%
mutate(targets = fct_inorder(targets)) %>%
ggplot(aes(w, fct_rev(targets), fill = H3K27ac)) +
geom_boxplot() +
scale_x_log10() +
scale_fill_scico_d(palette = "berlin")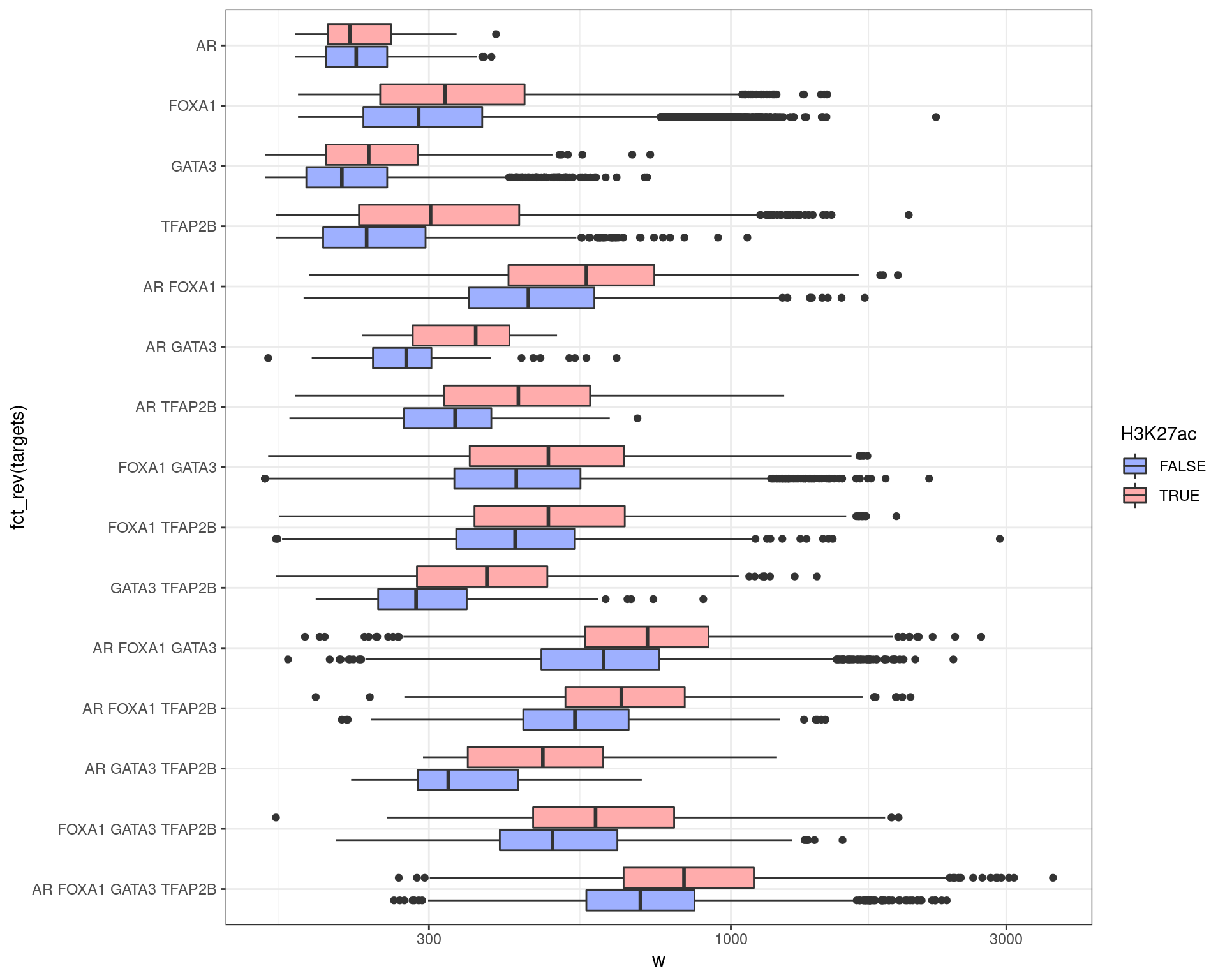
In most groups, wider peaks were more strongly associated with H3K27ac signal. Similarly, wider peaks were more commonly associated with multiple targets.
| Version | Author | Date |
|---|---|---|
| 1f959c3 | Steve Pederson | 2022-06-08 |
H3K27ac Signal
Given the hypothesis that the co-ocurrence of all 4 ChIP targets should be associated with increased transcriptional or regulatory activity, the set of all DHT peaks were then compared to promoters and enhancers derived H2K27ac binding, as detected in the existing GATA3/AR dataset produced by Leila Hosseinzadeh. Any H3K27ac peak detected in this dataset was classified either as a promoter or enhancer, and thus these features can be considered as the complete set of regions with detectable H3K27ac signal from this experiment.
fs <- 12
cp <- htmltools::tags$em(
glue(
"Summary of all sites, taking the union of all sites with any target bound. ",
"{comma(length(dht_consensus))} sites were found across all targets. ",
"Individual ChIP targets can be searched within this table using 't' or 'f' ",
"to denote True or False. Summaries regarding association with H3K27ac ",
"peaks and distance to the nearest TSS are also provided. ",
"The total numbers of sites overlapping H3K27ac-defined Promoters and ",
"Enhancers are shown with the % of sites being relative to to those ",
"overlapping H3K27ac-defined features"
)
)
tbl <- dht_consensus %>%
as_tibble() %>%
group_by(!!!syms(targets)) %>%
summarise(
n = dplyr::n(),
nH3K27ac = sum(H3K27ac),
H3K27ac = mean(H3K27ac),
promoter = sum(promoter),
enhancer = sum(enhancer),
d = median(dist_to_tss),
.groups = "drop"
) %>%
arrange(desc(n)) %T>%
write_tsv(
here::here("output/sites_by_h3k27ac.tsv")
) %>%
reactable(
filterable = TRUE,
pagination = FALSE,
columns = list(
AR = colDef(
cell = function(value) {
ifelse(value, "\u2714", "\u2716")
},
style = function(value) {
cl <- ifelse(value, "forestgreen", "red")
list(color = cl)
},
maxWidth = 60
),
FOXA1 = colDef(
cell = function(value) {
ifelse(value, "\u2714", "\u2716")
},
style = function(value) {
cl <- ifelse(value, "forestgreen", "red")
list(color = cl)
},
maxWidth = 60
),
GATA3 = colDef(
cell = function(value) {
ifelse(value, "\u2714", "\u2716")
},
style = function(value) {
cl <- ifelse(value, "forestgreen", "red")
list(color = cl)
},
maxWidth = 60
),
TFAP2B = colDef(
cell = function(value) {
ifelse(value, "\u2714", "\u2716")
},
style = function(value) {
cl <- ifelse(value, "forestgreen", "red")
list(color = cl)
},
maxWidth = 70
),
n = colDef(
name = "Nbr. Sites",
format = colFormat(separators = TRUE),
maxWidth = 75
),
nH3K27ac = colDef(
name = "Overlapping H3K27ac",
format = colFormat(separators = TRUE),
maxWidth = 85
),
H3K27ac = colDef(
name = "% H3K27ac Overlap",
format = colFormat(digits = 1, percent = TRUE),
style = function(value) {
bar_style(width = value, align = "right")
},
maxWidth = 80
),
promoter = colDef(
name = "Promoters",
style = function(value, index) {
bar_style(
width = 0.5*(value / .$nH3K27ac[[index]]), align = "right",
fill = rgb(1, 0.416, 0.416)
)
},
cell = function(value, index) {
lb <- glue("{value} ({percent(value / .$nH3K27ac[[index]], 1)})")
as.character(lb)
},
align = "left",
minWidth = 120
),
enhancer = colDef(
name = "Enhancers",
style = function(value, index) {
bar_style(
width = 0.5*(value / .$nH3K27ac[[index]]), align = "left",
fill = rgb(1, 0.65, 0.2)
)
},
cell = function(value, index) {
lb <- glue("{value} ({percent(value / .$nH3K27ac[[index]], 1)})")
as.character(lb)
},
align = "right",
minWidth = 120
),
d = colDef(
name = "Median Distance to TSS (bp)",
format = colFormat(digits = 0, separators = TRUE)
)
),
style = list(fontSize = fs)
)
div(class = "table",
div(class = "table-header",
div(class = "caption", cp),
tbl
)
)All H3K27ac Peaks
grp_h3k27ac <- dht_consensus %>%
as_tibble() %>%
dplyr::select(range, all_of(targets), H3K27ac) %>%
pivot_longer(cols = all_of(targets), names_to = "targets") %>%
dplyr::filter(value) %>%
group_by(range, H3K27ac) %>%
summarise(
targets = paste(targets, collapse = "+"),
.groups = "drop"
) %>%
mutate(width = width(GRanges(range)))
glm_h3k27ac <- glm(H3K27ac ~ 0 + targets + width, family = "binomial", data = grp_h3k27ac)A simple logistic regression model was then run to estimate the probability of overlapping a peak associated with the histone mark H3K27ac for each combination of ChIP targets. Binding of TFAP2\(\beta\) in isolation clearly had the strongest association with H3K27ac peaks, whilst the absence of TFAP2\(\beta\) clearly reduced the likelihood of all other ChIP targets being associated with H3K27ac signal.
inv.logit <- binomial()$linkinv
new_df <- grp_h3k27ac %>%
group_by(targets) %>%
summarise(width = median(width), .groups = "drop")
z <- qnorm(1 - 0.025 / nrow(new_df))
df <- predict(glm_h3k27ac, newdata = new_df, se.fit = TRUE) %>%
as_tibble() %>%
dplyr::select(ends_with("fit")) %>%
cbind(new_df) %>%
mutate(
lwr = inv.logit(fit - z*se.fit),
upr = inv.logit(fit + z*se.fit),
p = inv.logit(fit),
n_targets = str_count(targets, "\\+") + 1,
TFAP2B = str_detect(targets, "TFAP2B"),
targets = str_remove_all(targets, "\\+TFAP2B")
) %>%
arrange(n_targets, targets) %>%
mutate(
targets = fct_inorder(targets) %>% fct_rev(),
y = as.integer(targets) - 0.2 + 0.4*TFAP2B
)
df %>%
ggplot(aes(p, y, colour = TFAP2B)) +
geom_point() +
geom_errorbarh(aes(xmin = lwr, xmax = upr)) +
scale_y_continuous(
breaks = seq_along(levels(df$targets)),
labels = levels(df$targets),
expand = expansion(0.02)
) +
scale_x_continuous(
breaks = seq(0, 1, by = 0.2),
limits = c(0, 1), expand = expansion(c(0, 0))
) +
scale_colour_manual(values = c("grey30", "red")) +
labs(
x = "Probability of H3K27ac Peak Overlap",
y = "Detected Targets",
colour = "TFAP2B\nDetected"
) +
theme(
legend.position = c(0.99, 0.01),
legend.justification = c(1, 0)
)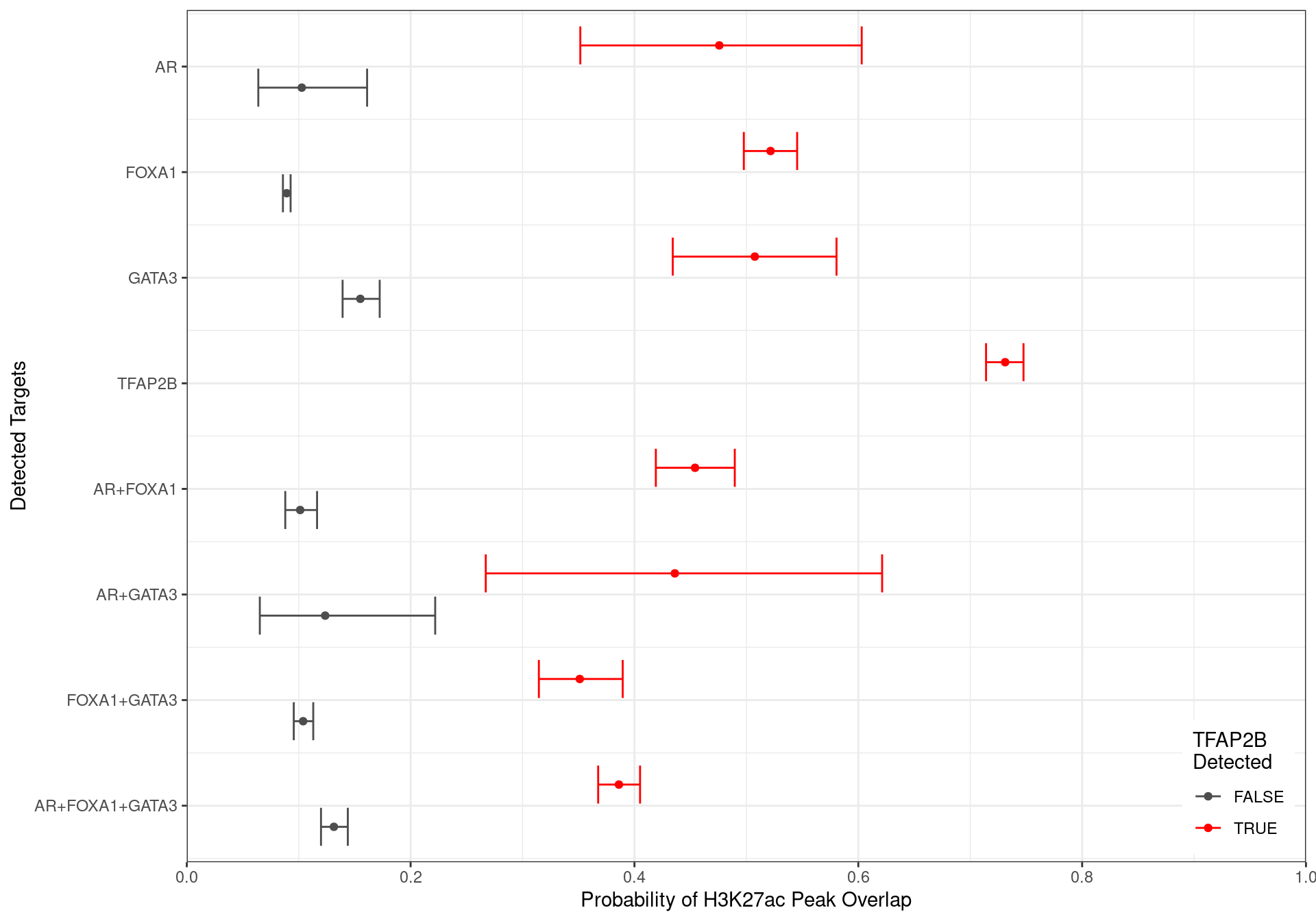
Family-wise 95% Confidence Intervals for the probability of overlapping an H3K27ac-derived feature, based on the combinations of detected ChIP targets. Given the impact peak width has on this probablity, intervals were generated holding width to be fixed at the median width for each group of targets.
Promoters Only
grp_prom <- dht_consensus %>%
as_tibble() %>%
dplyr::select(range, all_of(targets), promoter) %>%
pivot_longer(cols = all_of(targets), names_to = "targets") %>%
dplyr::filter(value) %>%
group_by(range, promoter) %>%
summarise(targets = paste(targets, collapse = "+"), .groups = "drop")%>%
mutate(width = width(GRanges(range)))
glm_prom <- glm(promoter ~ 0 + targets + width, family = "binomial", data = grp_prom)
df <- predict(glm_prom, newdata = new_df, se.fit = TRUE) %>%
as_tibble() %>%
dplyr::select(ends_with("fit")) %>%
cbind(new_df) %>%
mutate(
lwr = inv.logit(fit - z*se.fit),
upr = inv.logit(fit + z*se.fit),
p = inv.logit(fit),
n_targets = str_count(targets, "\\+") + 1,
TFAP2B = str_detect(targets, "TFAP2B"),
targets = str_remove_all(targets, "\\+TFAP2B")
) %>%
arrange(n_targets, targets) %>%
mutate(
targets = fct_inorder(targets) %>% fct_rev(),
y = as.integer(targets) - 0.2 + 0.4*TFAP2B
)
df %>%
ggplot(aes(p, y, colour = TFAP2B)) +
geom_point() +
geom_errorbarh(aes(xmin = lwr, xmax = upr)) +
scale_y_continuous(
breaks = seq_along(levels(df$targets)),
labels = levels(df$targets),
expand = expansion(0.02)
) +
scale_x_continuous(
breaks = seq(0, 1, by = 0.2),
limits = c(0, 1), expand = expansion(c(0, 0))
) +
scale_colour_manual(values = c("grey30", "red")) +
labs(
x = "Probability of Promoter Overlap",
y = "Detected Targets",
colour = "TFAP2B\nDetected"
) +
theme(
legend.position = c(0.99, 0.01),
legend.justification = c(1, 0)
)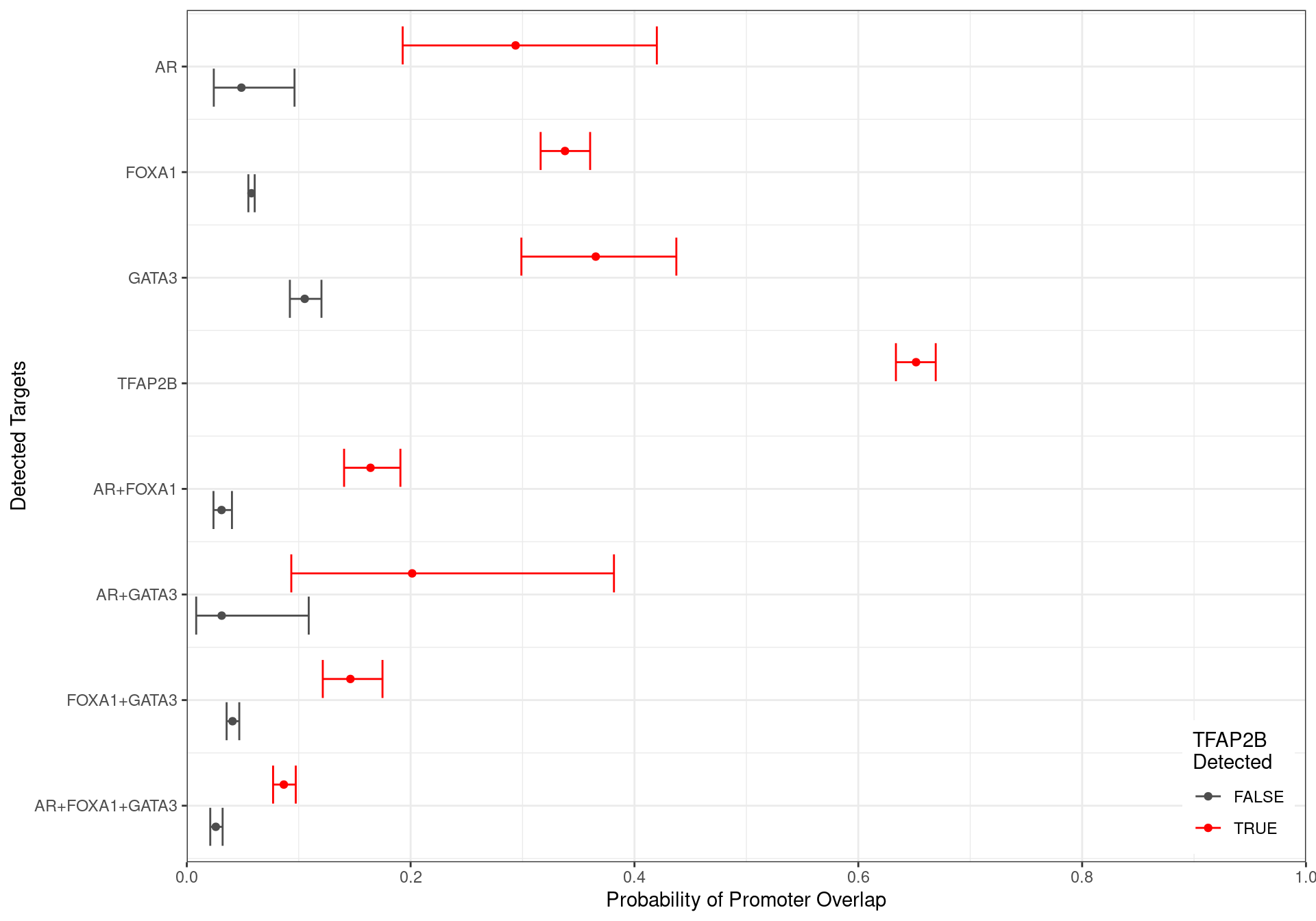
Family-wise 95% Confidence Intervals for the probability of overlapping an H3K27ac-derived promoter, based on the combinations of detected ChIP targets. Given the impact peak width has on this probablity, intervals were generated holding width to be fixed at the median width for each group of targets.
Enhancers Only
grp_enh <- dht_consensus %>%
as_tibble() %>%
dplyr::select(range, all_of(targets), enhancer) %>%
pivot_longer(cols = all_of(targets), names_to = "targets") %>%
dplyr::filter(value) %>%
group_by(range, enhancer) %>%
summarise(targets = paste(targets, collapse = "+"), .groups = "drop") %>%
mutate(width = width(GRanges(range)))
glm_enh <- glm(enhancer ~ 0 + targets + width, family = "binomial", data = grp_enh)
df <- predict(glm_enh, newdata = new_df, se.fit = TRUE) %>%
as_tibble() %>%
dplyr::select(ends_with("fit")) %>%
cbind(new_df) %>%
mutate(
lwr = inv.logit(fit - z*se.fit),
upr = inv.logit(fit + z*se.fit),
p = inv.logit(fit),
n_targets = str_count(targets, "\\+") + 1,
TFAP2B = str_detect(targets, "TFAP2B"),
targets = str_remove_all(targets, "\\+TFAP2B")
) %>%
arrange(n_targets, targets) %>%
mutate(
targets = fct_inorder(targets) %>% fct_rev(),
y = as.integer(targets) - 0.2 + 0.4*TFAP2B
)
df %>%
ggplot(aes(p, y, colour = TFAP2B)) +
geom_point() +
geom_errorbarh(aes(xmin = lwr, xmax = upr)) +
scale_y_continuous(
breaks = seq_along(levels(df$targets)),
labels = levels(df$targets),
expand = expansion(0.02)
) +
scale_x_continuous(
breaks = seq(0, 1, by = 0.2),
limits = c(0, 1), expand = expansion(c(0, 0))
) +
scale_colour_manual(values = c("grey30", "red")) +
labs(
x = "Probability of Enhancer Overlap",
y = "Detected Targets",
colour = "TFAP2B\nDetected"
) +
theme(
legend.position = c(0.99, 0.01),
legend.justification = c(1, 0)
)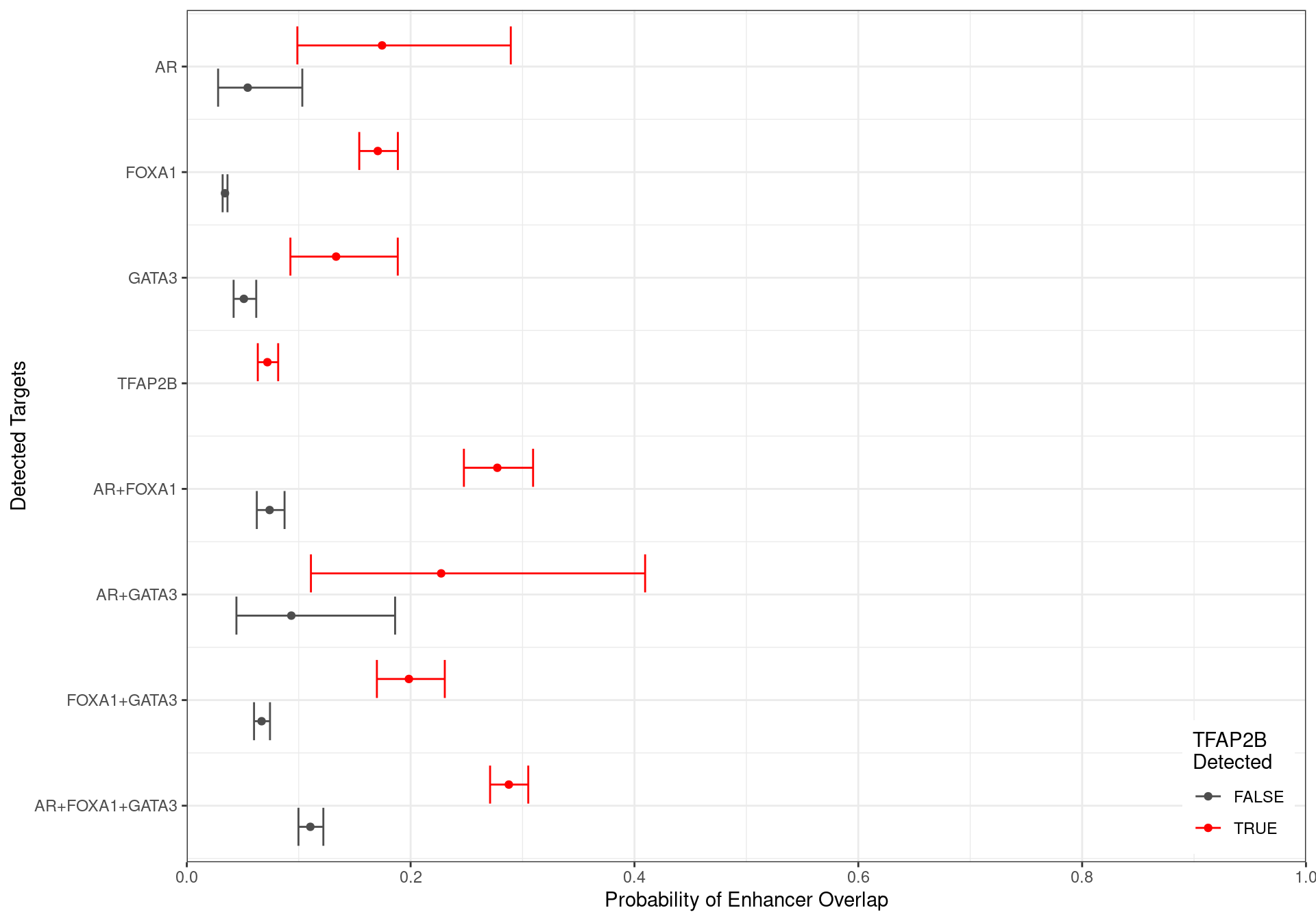
Family-wise 95% Confidence Intervals for the probability of overlapping an H3K27ac-derived enhancer, based on the combinations of detected ChIP targets. Given the impact peak width has on this probablity, intervals were generated holding width to be fixed at the median width for each group of targets.
Distance to Nearest TSS
The distance to the nearest TSS was also checked for each of the union peaks, with the vast majority of TFAP2\(\beta\)-only peaks being with 1kb of a TSS, indicating a fundamental role for TFAP2\(\beta\) in transcriptional regulation. These were much further away when bound in combination with other ChIP targets, to the point that most sites where all four were found bound were >3kb from any TSS, suggesting that an alternative role for TFAP2\(\beta\) is enhancer-based gene regulation, when acting in concert with the remaining targets.
df <- dht_consensus %>%
as_tibble() %>%
dplyr::select(range, all_of(targets), dist_to_tss, H3K27ac) %>%
pivot_longer(cols = all_of(targets), names_to = "targets") %>%
dplyr::filter(value) %>%
group_by(range, dist_to_tss, H3K27ac) %>%
arrange(range, targets) %>%
summarise(targets = paste(targets, collapse = "+"), .groups = "drop") %>%
mutate(
n_targets = str_count(targets, "\\+") + 1,
TFAP2B = ifelse(str_detect(targets, "TFAP2B"), "TFAP2B", "No TFAP2B")
) %>%
arrange(desc(n_targets), desc(targets)) %>%
mutate(
targets = fct_inorder(targets) %>%
fct_relabel(str_remove_all, pattern = "\\+TFAP2B")
) %>%
group_by(targets, TFAP2B, H3K27ac) %>%
mutate(
q = rank(dist_to_tss, ties.method = "first") / dplyr::n()
) %>%
ungroup() %>%
dplyr::filter(dist_to_tss < 5e3)
brk <- c(0, 100, 2e3, Inf)
lb <- paste(c("Core (<100)", "Distal (<2kb)"), "Promoter") %>%
c("Enhancer")
a <- df %>%
mutate(
region = dist_to_tss %>%
cut(
breaks = brk, include.lowest = TRUE, labels = lb
)
) %>%
group_by(targets, TFAP2B, H3K27ac, region) %>%
summarise(n = dplyr::n(), .groups = "drop_last") %>%
mutate(prop = n / sum(n)) %>%
ungroup() %>%
mutate(
status = ifelse(
H3K27ac,
paste("Active -", TFAP2B),
paste("Inactive -", TFAP2B)
) %>%
factor(
levels = c(
"Active - TFAP2B", "Active - No TFAP2B",
"Inactive - TFAP2B", "Inactive - No TFAP2B"
)
)
) %>%
ggplot(aes(prop, fct_rev(status), fill = fct_rev(region))) +
geom_col() +
facet_wrap(~fct_rev(targets), ncol = 4) +
scale_x_continuous(labels = percent) +
scale_fill_viridis_d(direction = -1) +
labs(
x = "Percent (%)",
y = "H3K27ac Marks + TFAP2B Status",
fill = "Regulatory Region"
) +
theme(legend.position = "bottom")
b <- df %>%
mutate(
region = dist_to_tss %>%
cut(
breaks = brk, include.lowest = TRUE, labels = lb
)
) %>%
group_by(targets, TFAP2B, H3K27ac, region) %>%
summarise(n = dplyr::n(), .groups = "drop_last") %>%
mutate(prop = n / sum(n)) %>%
ungroup() %>%
mutate(
status = ifelse(
H3K27ac,
paste("Active -", TFAP2B),
paste("Inactive -", TFAP2B)
) %>%
factor(
levels = c(
"Active - TFAP2B", "Active - No TFAP2B",
"Inactive - TFAP2B", "Inactive - No TFAP2B"
)
)
) %>%
ggplot(aes(status, n, fill = fct_rev(region))) +
geom_col() +
facet_wrap(~fct_rev(targets), ncol = 4) +
scale_y_continuous(labels = comma) +
scale_fill_viridis_d(direction = -1) +
labs(
y = "Number of Peaks",
x = "H3K27ac Marks + TFAP2B Status",
fill = "Regulatory Region"
) +
theme(
legend.position = "none",
axis.text.x = element_text(angle = 90, hjust = 1, vjust = 1/2)
)
plot_grid(
a,
b,
labels = c("A", "B"),
nrow = 2
# rel_widths = c(0.5, 0.5)
)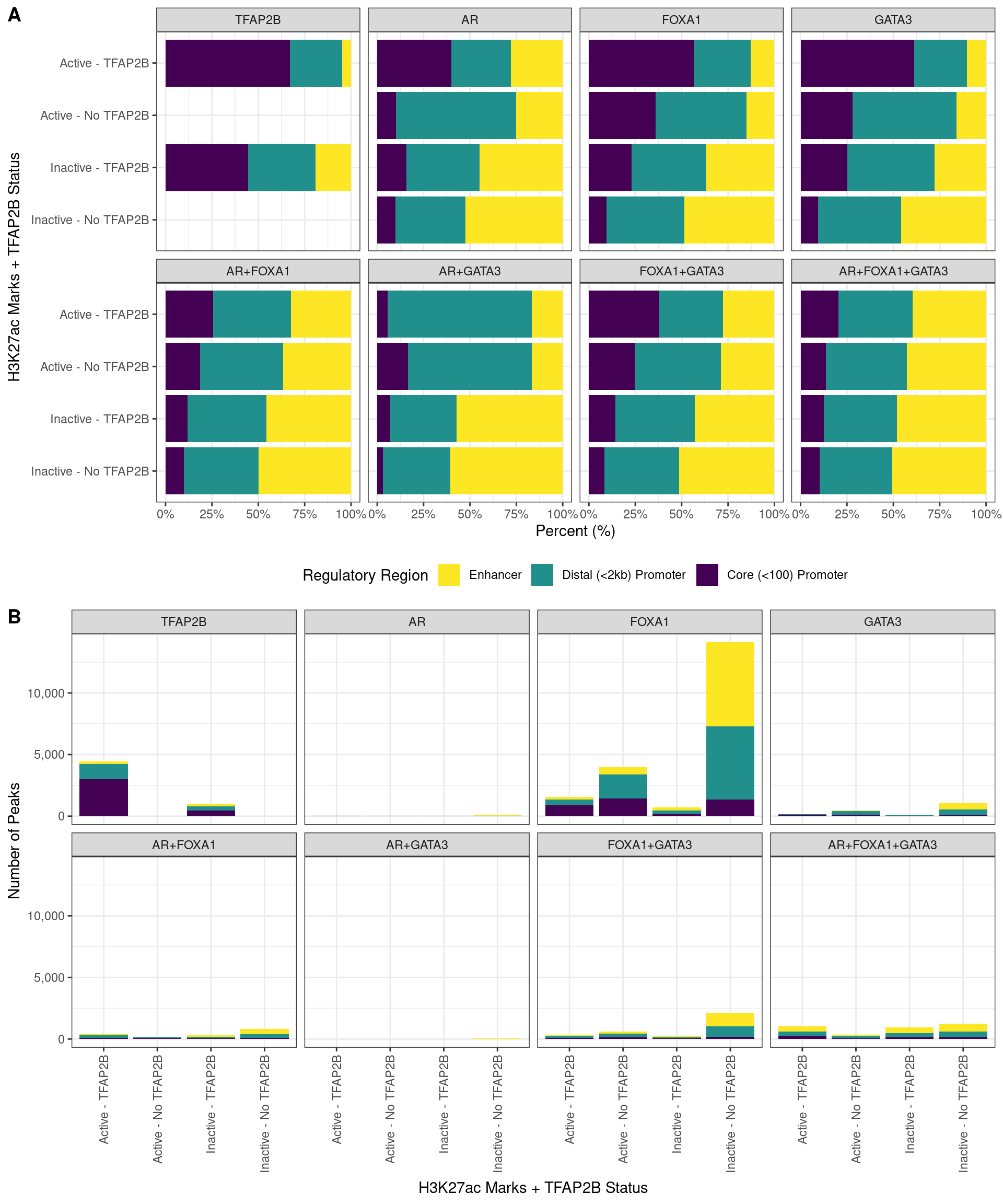
Breaking peak overlaps into overlaps with the Core Promoter (<100bp from TSS), Distal Promoter (<2kb from TSS) and Enhancers (>2kb from TSS), the presence of TFAP2B showed in increased affinity for promoters, particularly when only binding with a single additional factor. Similarly, peaks marked as active by an overlap with H3K27ac signal were more strongly associated with promoters. Of the H3K27ac-marked peaks where TFAP2B was detected, the largets proportion which bound enhancers, was found where all three remaining targets were also detected, indicating an increased preference for enhancers with this bindin combination. Union peaks are shown A) as percentages of peaks which overlap a given region, or B) the total number of peaks which overlap a given region. The most common binding event in the dataset was clearly FOXA1 in isolation, binding regions which are not marked by H3K27ac and do not also bind TFAP2B. The next most common set of peaks was those binding FOXA1 alone in an H3K27ac marked region, followed by TFAP2B in an H3K27ac marked region.
| Version | Author | Date |
|---|---|---|
| 1f959c3 | Steve Pederson | 2022-06-08 |
Comparison With H3K27ac HiChIP Data
fl <- here::here("data", "hichip") %>%
list.files(full.names = TRUE, pattern = "gi.+rds")
hic <- GInteractions()
for (f in fl) {
hic <- c(hic, read_rds(f))
}
hic <- sort(hic)H3K27ac-HiChIP obtained from SRA and analysed previously, was additionally included to improve mapping of peaks to genes. Whilst the previous H3K27ac-derived features were obtained from the same passages/experiments as GATA3 and AR, HiChIP data was obtained from a public dataset, not produced with the DRMCRL, and were defined using only the Vehicle controls from an Abemaciclib Vs. Vehicle experiment. Long range interactions were included for bin sizes of 5, 10, 20 and 40 kb. Before proceeding, the comparability of the H3K27ac-HiChIP and H3K27ac-derived features was checked. 93% of HiChIP long-range interactions overlapped a promoter or enhancer derived from H3K27ac ChIP-seq. Conversely, 86% or ChIP-Seq features mapped to a long-range interaction, confirming that the H3K27ac signatures within MDA-MB-453 cells are highly comparable across laboratories.
Comparison To Key Genes
data("grch37.cytobands")
tm <- read_rds(
here::here("data", "annotations", "trans_models.rds")
)
bwfl <- list(
AR = here::here("data", "bigwig", "AR") %>%
list.files(pattern = "DHT_merged.+bw", full.names = TRUE) %>%
BigWigFileList() %>%
setNames(
path(.) %>%
basename() %>%
str_remove_all("_merged_treat.+")
),
FOXA1 = here::here("data", "bigwig", "FOXA1") %>%
list.files(pattern = "DHT_merged.+bw", full.names = TRUE) %>%
BigWigFileList() %>%
setNames(
path(.) %>%
basename() %>%
str_remove_all("_merged_treat.+")
),
GATA3 = here::here("data", "bigwig", "GATA3") %>%
list.files(pattern = "DHT_merged.+bw", full.names = TRUE) %>%
BigWigFileList() %>%
setNames(
path(.) %>%
basename() %>%
str_remove_all("_merged_treat.+")
),
TFAP2B = here::here("data", "bigwig", "TFAP2B") %>%
list.files(pattern = "DHT_merged.+bw", full.names = TRUE) %>%
BigWigFileList() %>%
setNames(
path(.) %>%
basename() %>%
str_remove_all("_merged_treat.+")
),
H3K27ac = here::here("data", "bigwig", "H3K27ac") %>%
list.files(pattern = "merged_DHT.bw", full.names = TRUE) %>%
BigWigFileList() %>%
setNames(
path(.) %>%
basename() %>%
str_extract("DHT|Veh")
)
) %>%
lapply(path) %>%
lapply(as.character) %>%
BigWigFileList()
y_lim <- here::here("data", "bigwig", targets) %>%
sapply(
list.files, pattern = "merged.*summary", full.names= TRUE, simplify = FALSE
) %>%
lapply(
function(x) lapply(x, read_tsv)
) %>%
lapply(bind_rows) %>%
lapply(function(x) c(0, max(x$score))) %>%
setNames(basename(names(.)))
y_lim$H3K27ac <- bwfl$H3K27ac %>%
import.bw %>%
mcols() %>%
.[["score"]] %>%
max() %>%
c(0) %>%
range()Mapping To Detected Genes
rnaseq <- here::here("data", "rnaseq", "dge.rds") %>%
read_rds()
counts <- here::here("data", "rnaseq", "counts.out.gz") %>%
read_tsv(skip = 1) %>%
dplyr::select(Geneid, ends_with("bam"))
detected <- counts %>%
pivot_longer(
cols = ends_with("bam"), names_to = "sample", values_to = "counts"
) %>%
mutate(detected = counts > 0) %>%
group_by(Geneid) %>%
summarise(detected = mean(detected) > 0.25, .groups = "drop") %>%
dplyr::filter(detected) %>%
pull("Geneid")
top_table <- here::here("data", "rnaseq", "MDA-MB-453_RNASeq.tsv") %>%
read_tsv() # Not a DHT-treatment analysis though...dht_consensus <- mapByFeature(
dht_consensus,
genes = subset(all_gr$gene, gene_id %in% detected),
prom = subset(features, feature == "promoter"),
## Given we have accuracte H3K27ac HiChIP, better mappings can be gained that way.
## Exclude any enhancers which overlap the HiChIP
enh = subset(features, feature == "enhancer") %>% filter_by_non_overlaps(hic),
gi = hic
) %>%
mutate(mapped = vapply(gene_id, function(x) length(x) > 0, logical(1)))
all_targets <- dht_consensus %>%
as_tibble() %>%
dplyr::filter(if_all(all_of(targets))) %>%
unnest(everything()) %>%
distinct(gene_id) %>%
pull("gene_id")In order to more accurately assign genes to actively transcribed genes, the RNA-Seq dataset generated in 2013 studying DHT Vs. Vehicle in MDA-MB-453 cells was used to define the set of genes able to be considered as detected. As this was a polyA dataset, some ncRNA which are not adenylated may be expressed, but will remain as undetected despite this technological limitation. All 21,328 genes with >1 read in at least 1/4 of the samples was considered to be detected, and peaks were only mapped to detected genes.
Mapping was performed using mapByFeature() from the
package extraChIP using:
- Promoters defined externally using H3K27ac
- Only the externally defined enhancers which contained no H3K27ac-HiChIP overlap
- H3K27ac HiChIP interactions for long range mapping
No separate enhancers were using in the mapping steps as the
H3K27ac-HiChIP interaction were considered to provide clear associations
between peaks and target genes. By contrast, the default mapping of
mapByFeature() would map all genes within 100kb to any peak
considered as an enhancer
90% of detected genes had one or more peaks mapped to them.
Looking specifically at the peaks for which all four targets directly overlapped, 10,187 of the 21,328 detected genes were mapped to at least one directly overlapping binding region.
Apocrine Signature Enrichment
Subtype Gene Signatures
hgnc <- read_csv(
here::here("data", "external", "hgnc-symbol-check.csv"), skip = 1
) %>%
dplyr::select(Gene = Input, gene_name = `Approved symbol`)
apo_ranks <- here::here("data", "external", "ApoGenes.txt") %>%
read_tsv() %>%
left_join(hgnc) %>%
left_join(
as_tibble(all_gr$gene) %>%
dplyr::select(gene_id, gene_name),
by = "gene_name"
) %>%
dplyr::select(starts_with("gene_"), ends_with("rank")) %>%
dplyr::filter(!is.na(gene_id))
apo_full <- read_excel(
here::here("data", "external", "Apocrine gene lists.xls"),
col_names = c(
"Gene",
paste0("ESR1", c("_logFC", "_p")),
paste0("CLCA2", c("_logFC", "_p")),
paste0("DCN", c("_logFC", "_p")),
paste0("GZMA", c("_logFC", "_p")),
paste0("MX1", c("_logFC", "_p")),
paste0("TPX2", c("_logFC", "_p")),
paste0("FABP4", c("_logFC", "_p")),
paste0("ADM", c("_logFC", "_p")),
paste0("CD83", c("_logFC", "_p"))
),
col_types = c("text", rep("numeric", 18)),
skip = 1
) %>%
left_join(hgnc) %>%
left_join(
as_tibble(all_gr$gene) %>%
dplyr::select(gene_id, gene_name),
by = "gene_name"
) %>%
dplyr::select(starts_with("gene", FALSE), matches("_(logFC|p)"))
apo_genesets <- colnames(apo_full) %>%
str_subset("_p$") %>%
sapply(
function(x) {
dplyr::filter(apo_full, !!sym(x) < 0.05, !is.na(gene_name))$gene_name
},
simplify = FALSE
) %>%
setNames(
str_remove(names(.), pattern = "_p")
)
genes_by_subtype <- apo_genesets %>%
lapply(list) %>%
as_tibble() %>%
pivot_longer(everything(), names_to = "subtype", values_to = "gene_name") %>%
unnest(everything()) %>%
split(.$gene_name) %>%
lapply(pull, "subtype")The models for different subtypes, as previously published were loaded, with a signature from each subtype being derived from genes with an adjusted p-value < 0.05, as initially published. This gave 9 different gene-sets with key marker genes denoting the subtypes: ESR1, CLCA2, DCN, GZMA, MX1, TPX2, FABP4, ADM and CD83
apo_genesets %>%
fromList() %>%
upset(
sets = rev(names(.)),
# keep.order = TRUE,
order.by = "freq",
set_size.show = TRUE,
set_size.scale_max = nrow(.) * 0.4
)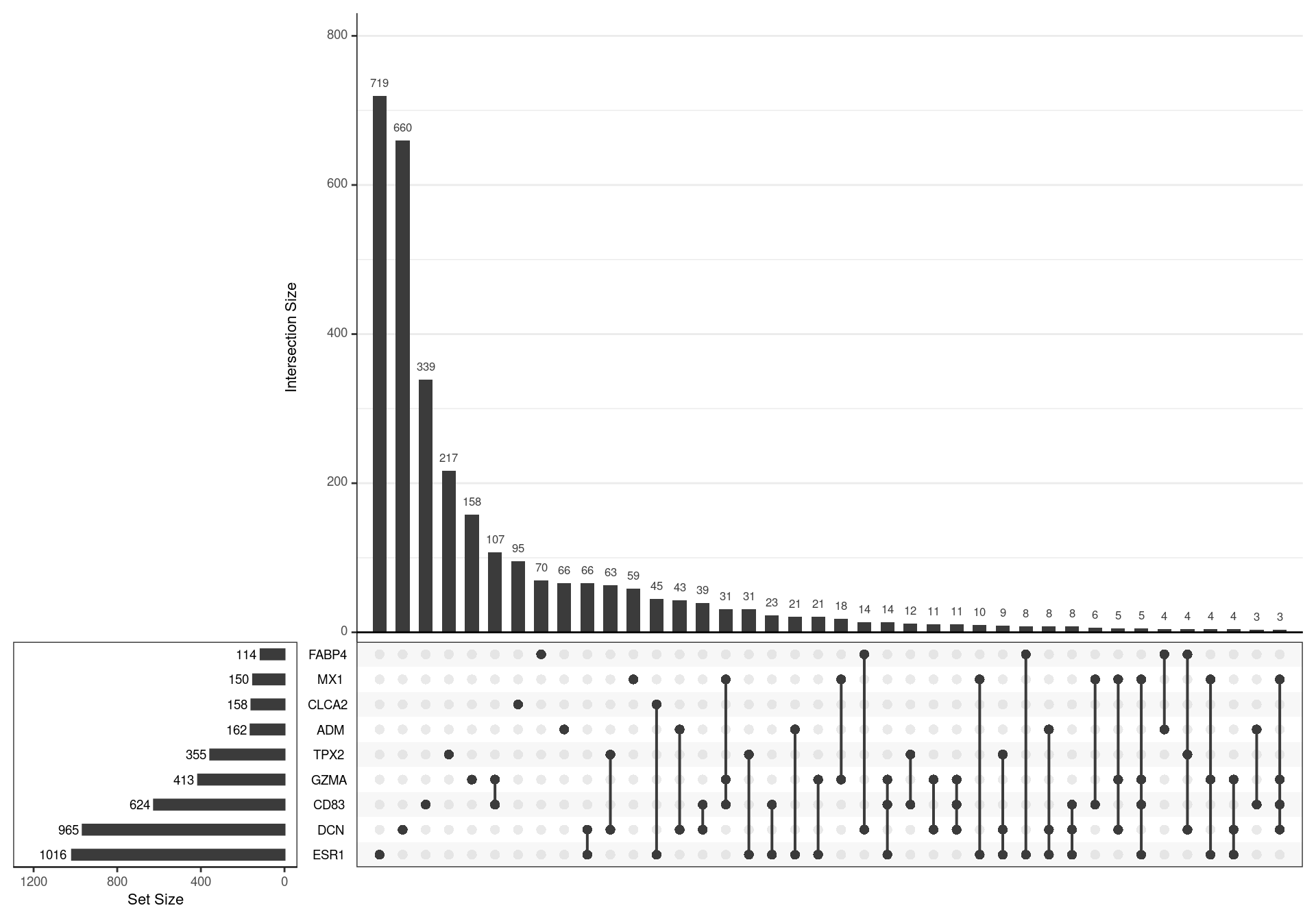
UpSet plot showing the association between genes contained in each signature. A relatively high degree of similarity was noted between some signature pairs, such as GZMA/CD83 and MX1/GZMA
apo_genesets %>%
fromList() %>%
cor() %>%
corrplot(
diag = FALSE,
addCoef.col = "grey50",
addCoefasPercent = TRUE
)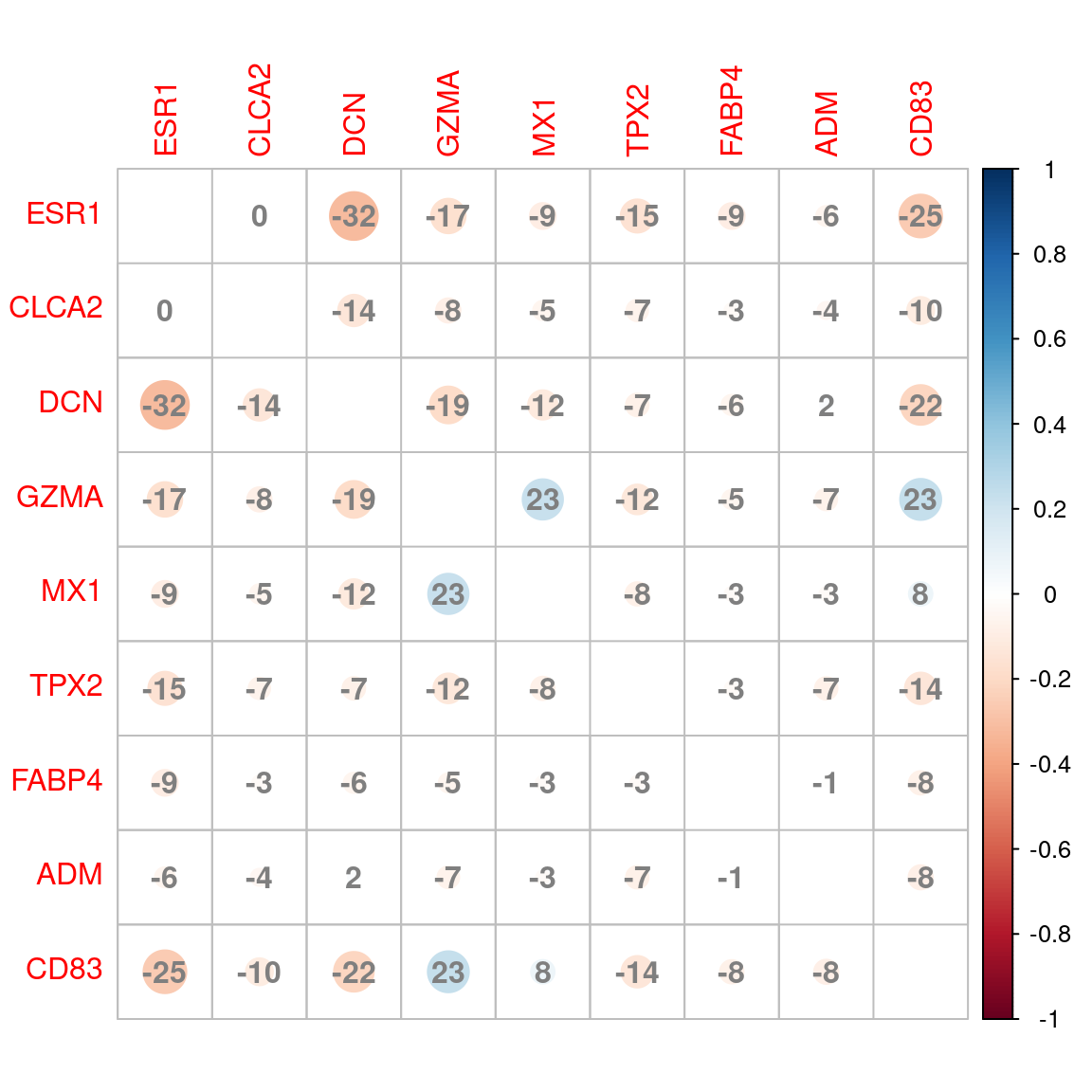
Correlation plot between signatures based on shared genes between groups
Enrichment Testing
The set of genes mapped to sites contain all four ChIP targets were
then tested for enrichment of any of the defined signatures. Enrichment
testing as implemented in goseq was used, incorporating the
number of consensus peaks mapped to a gene as the offset term for biased
sampling.
All Common Sites
mapped <- all_gr$gene %>%
filter(gene_id %in% detected) %>%
mutate(gene_width = width) %>%
as_tibble() %>%
dplyr::select(starts_with("gene")) %>%
mutate(
mapped_to_all = gene_name %in% unlist(filter(dht_consensus, !!!syms(targets))$gene_name),
n_peaks = table(unlist(dht_consensus$gene_name))[gene_name] %>%
as.integer(),
n_peaks = ifelse(is.na(n_peaks), 0, n_peaks)
) %>%
distinct(gene_name, .keep_all = TRUE)
goseq_all4 <- structure(
mapped$mapped_to_all, names = mapped$gene_name
) %>%
nullp(bias.data = mapped$n_peaks) %>%
goseq(gene2cat = genes_by_subtype) %>%
as_tibble() %>%
dplyr::select(
subtype = category,
p = over_represented_pvalue,
n_mapped = numDEInCat,
signature_size = numInCat
) %>%
mutate(fdr = p.adjust(p, "fdr"))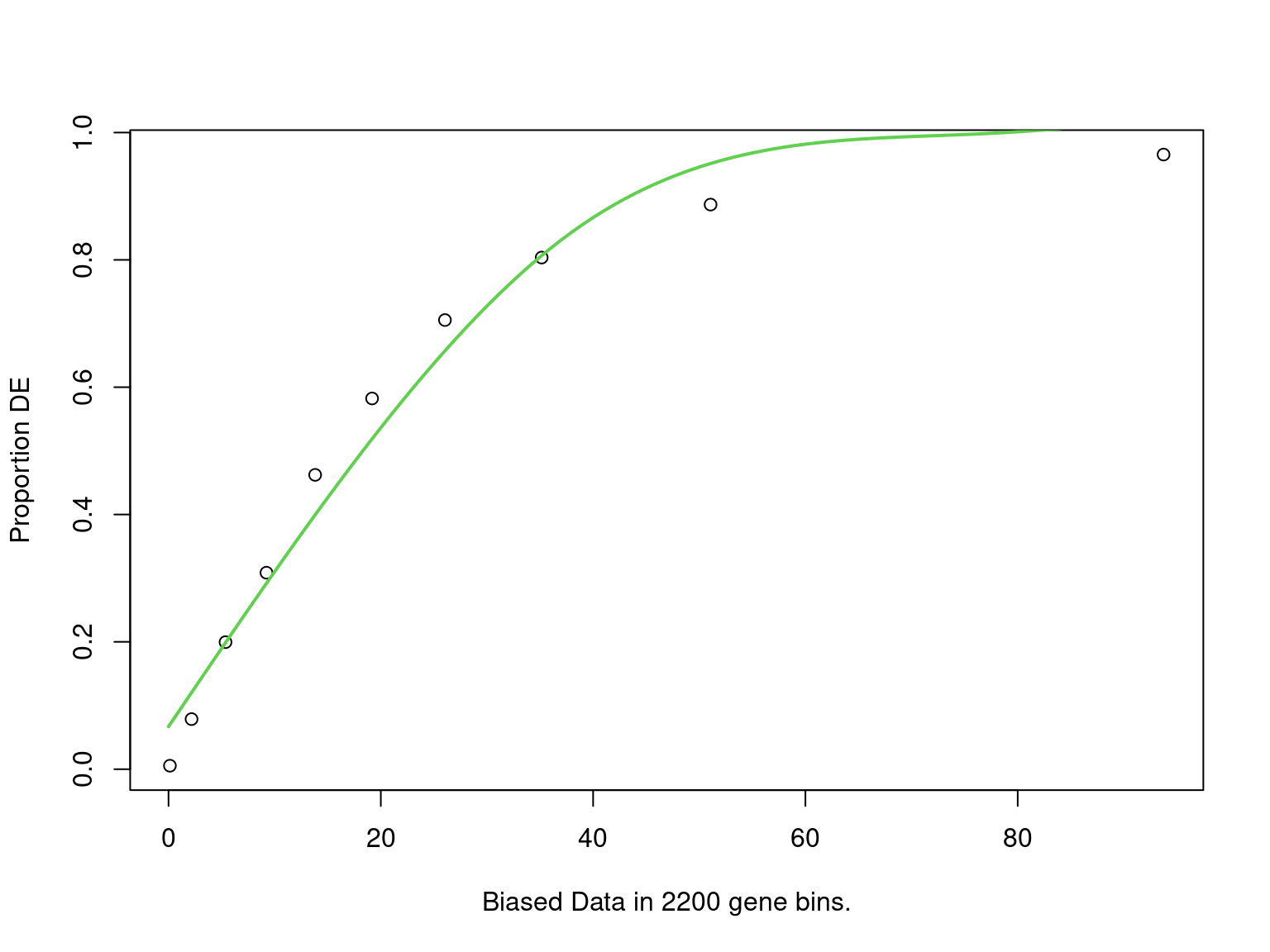
PWF for biased sampling using the number of peaks mapped to a gene as the bias offset
goseq_all4 %>%
mutate(`%` = percent(n_mapped / signature_size, accuracy = 0.1)) %>%
dplyr::select(
Subtype = subtype, `Signature Size` = signature_size, `# Mapped` = n_mapped,
`% Mapped` = `%`, p, FDR = fdr
) %>%
pander(
justify = "lrrrrr",
caption = paste(
"Results for Subtype enrichment testing using all genes mapped to any ",
"consensus peak, where all four ChIP targets were considered to be present."
),
emphasize.strong.rows = which(.$FDR < 0.05)
)| Subtype | Signature Size | # Mapped | % Mapped | p | FDR |
|---|---|---|---|---|---|
| CLCA2 | 136 | 104 | 76.5% | 0.0007285 | 0.006557 |
| ESR1 | 868 | 535 | 61.6% | 0.05081 | 0.2286 |
| ADM | 123 | 74 | 60.2% | 0.1779 | 0.5336 |
| TPX2 | 333 | 200 | 60.1% | 0.5204 | 0.946 |
| DCN | 766 | 435 | 56.8% | 0.8338 | 0.946 |
| CD83 | 400 | 210 | 52.5% | 0.9036 | 0.946 |
| GZMA | 195 | 97 | 49.7% | 0.9104 | 0.946 |
| MX1 | 106 | 47 | 44.3% | 0.9372 | 0.946 |
| FABP4 | 50 | 21 | 42.0% | 0.946 | 0.946 |
H3K27Ac-Associated Common Sites
The same process was then performed restricting the mappings to those sites associated with H3K27ac binding.
mapped_h3k <- all_gr$gene %>%
filter(gene_id %in% detected) %>%
mutate(gene_width = width) %>%
as_tibble() %>%
dplyr::select(starts_with("gene")) %>%
mutate(
mapped_to_all = gene_name %in% unlist(filter(dht_consensus, !!!syms(targets), H3K27ac)$gene_name),
n_peaks = table(unlist(dht_consensus$gene_name))[gene_name] %>%
as.integer(),
n_peaks = ifelse(is.na(n_peaks), 0, n_peaks)
) %>%
distinct(gene_name, .keep_all = TRUE)
goseq_h3k <- structure(
mapped_h3k$mapped_to_all, names = mapped_h3k$gene_name
) %>%
nullp(bias.data = mapped_h3k$n_peaks) %>%
goseq(gene2cat = genes_by_subtype) %>%
as_tibble() %>%
dplyr::select(
subtype = category,
p = over_represented_pvalue,
n_mapped = numDEInCat,
signature_size = numInCat
) %>%
mutate(fdr = p.adjust(p, "fdr"))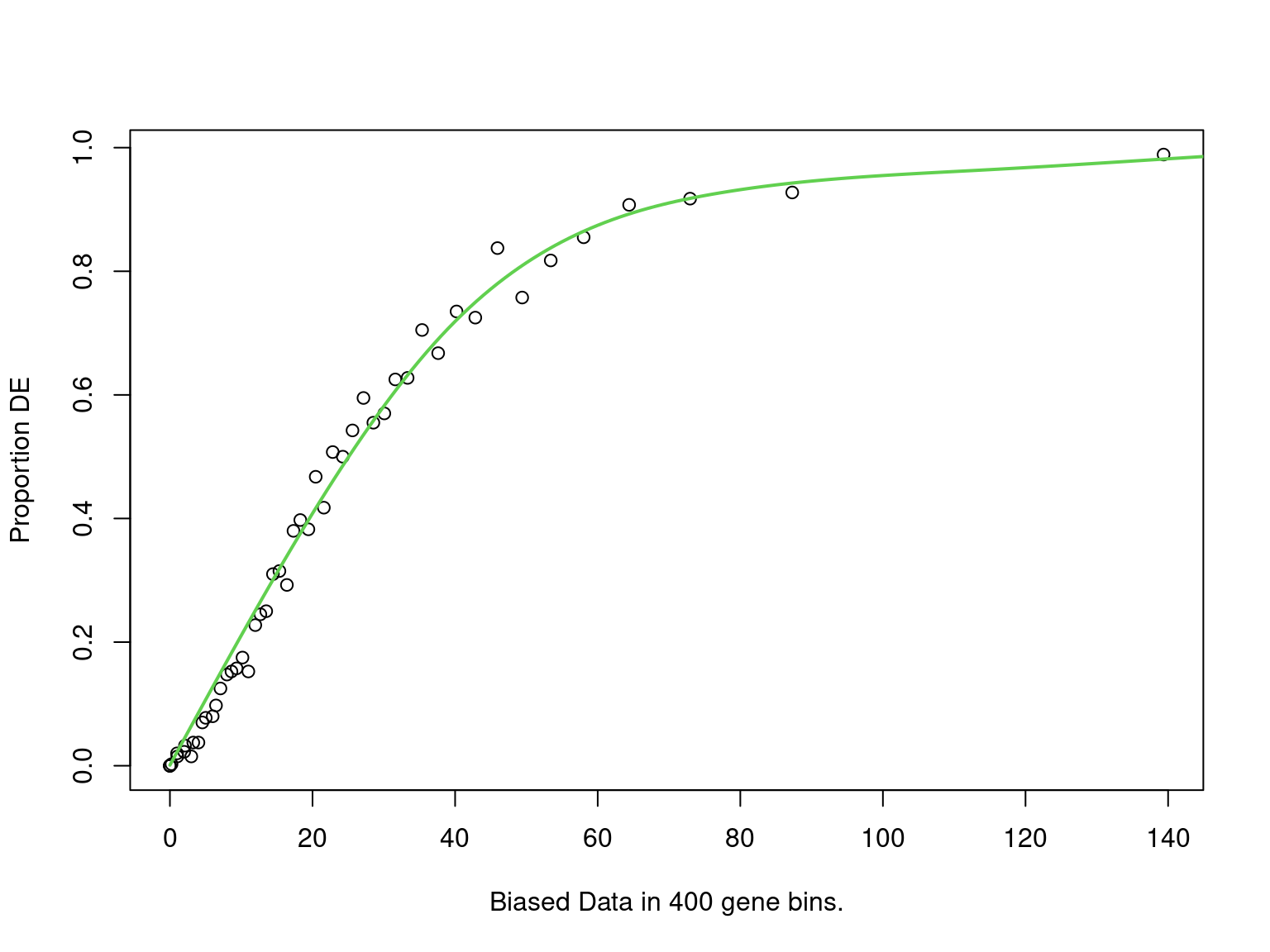
PWF for biased sampling using the number of peaks mapped to a gene as the bias offset
goseq_h3k %>%
mutate(`%` = percent(n_mapped / signature_size, accuracy = 0.1)) %>%
dplyr::select(
Subtype = subtype, `Signature Size` = signature_size, `# Mapped` = n_mapped,
`% Mapped` = `%`, p, FDR = fdr
) %>%
pander(
justify = "lrrrrr",
caption = paste(
"Results for Subtype enrichment testing using only genes mapped to a",
"consensus peak *associated with H3K27ac activity*, and where all four",
"ChIP targets were considered to be present."
),
emphasize.strong.rows = which(.$FDR < 0.05)
)| Subtype | Signature Size | # Mapped | % Mapped | p | FDR |
|---|---|---|---|---|---|
| CLCA2 | 136 | 91 | 66.9% | 5.577e-05 | 0.0005019 |
| ESR1 | 868 | 430 | 49.5% | 0.003229 | 0.01453 |
| ADM | 123 | 55 | 44.7% | 0.2842 | 0.8527 |
| FABP4 | 50 | 17 | 34.0% | 0.7642 | 0.9859 |
| MX1 | 106 | 36 | 34.0% | 0.7944 | 0.9859 |
| TPX2 | 333 | 147 | 44.1% | 0.8391 | 0.9859 |
| GZMA | 195 | 71 | 36.4% | 0.8773 | 0.9859 |
| CD83 | 400 | 147 | 36.8% | 0.9852 | 0.9859 |
| DCN | 766 | 315 | 41.1% | 0.9859 | 0.9859 |
H3K27ac Associated Sites Without TFAP2B
The same process was then performed restricting the mappings to those sites associated with H3K27ac binding, where AR, FOXA1 and GATA3 are all bound but without TFAP2B.
mapped_noap2b <- all_gr$gene %>%
filter(gene_id %in% detected) %>%
mutate(gene_width = width) %>%
as_tibble() %>%
dplyr::select(starts_with("gene")) %>%
mutate(
mapped_to_all = gene_name %in% unlist(filter(dht_consensus, AR, FOXA1, GATA3, !TFAP2B, H3K27ac)$gene_name),
n_peaks = table(unlist(dht_consensus$gene_name))[gene_name] %>%
as.integer(),
n_peaks = ifelse(is.na(n_peaks), 0, n_peaks)
) %>%
distinct(gene_name, .keep_all = TRUE)
goseq_noap2b <- structure(
mapped_noap2b$mapped_to_all, names = mapped_noap2b$gene_name
) %>%
nullp(bias.data = mapped_noap2b$n_peaks) %>%
goseq(gene2cat = genes_by_subtype) %>%
as_tibble() %>%
dplyr::select(
subtype = category,
p = over_represented_pvalue,
n_mapped = numDEInCat,
signature_size = numInCat
) %>%
mutate(fdr = p.adjust(p, "fdr"))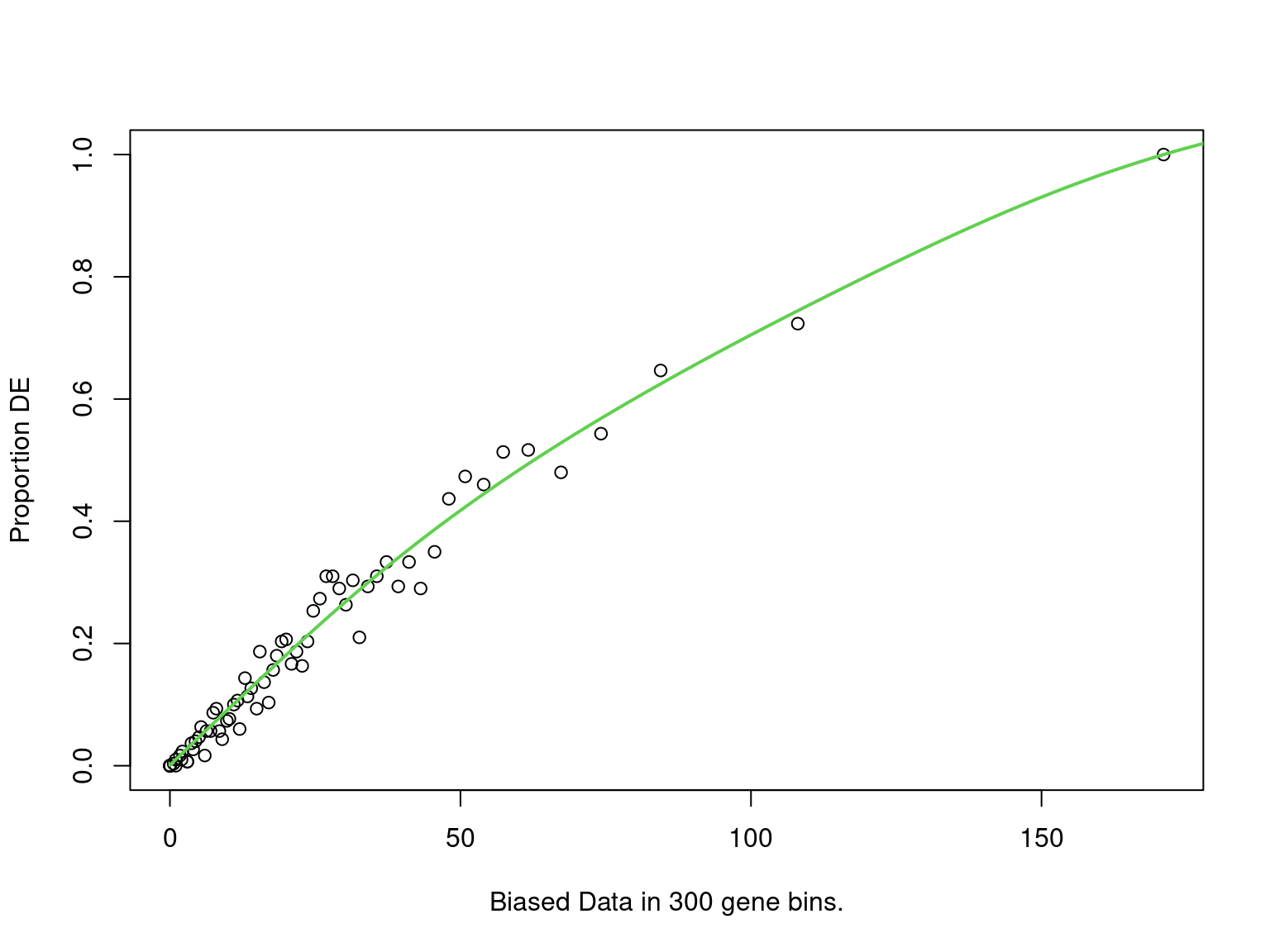
PWF for biased sampling using the number of peaks mapped to a gene as the bias offset
goseq_noap2b %>%
mutate(`%` = percent(n_mapped / signature_size, accuracy = 0.1)) %>%
dplyr::select(
Subtype = subtype, `Signature Size` = signature_size, `# Mapped` = n_mapped,
`% Mapped` = `%`, p, FDR = fdr
) %>%
pander(
justify = "lrrrrr",
caption = paste(
"Results for Subtype enrichment testing using only genes mapped to a",
"consensus peak *associated with H3K27ac activity*, and where AR, FOXA1",
"and GATA3 were considered to be present, but in the absence of TFAP2B."
),
emphasize.strong.rows = which(.$FDR < 0.05)
)| Subtype | Signature Size | # Mapped | % Mapped | p | FDR |
|---|---|---|---|---|---|
| CLCA2 | 136 | 46 | 33.8% | 0.05437 | 0.417 |
| ESR1 | 868 | 214 | 24.7% | 0.09266 | 0.417 |
| DCN | 766 | 168 | 21.9% | 0.4691 | 0.8998 |
| ADM | 123 | 26 | 21.1% | 0.4962 | 0.8998 |
| TPX2 | 333 | 79 | 23.7% | 0.4999 | 0.8998 |
| FABP4 | 50 | 7 | 14.0% | 0.7896 | 0.9987 |
| MX1 | 106 | 16 | 15.1% | 0.7964 | 0.9987 |
| GZMA | 195 | 29 | 14.9% | 0.943 | 0.9987 |
| CD83 | 400 | 61 | 15.2% | 0.9987 | 0.9987 |
H3K27ac Associated Sites Without AR
The same process was then performed restricting the mappings to those sites associated with H3K27ac binding, where FOXA1, GATA3 and TFAP2B are all bound but without taking AR binding into account.
mapped_noar <- all_gr$gene %>%
filter(gene_id %in% detected) %>%
mutate(gene_width = width) %>%
as_tibble() %>%
dplyr::select(starts_with("gene")) %>%
mutate(
mapped_to_all = gene_name %in% unlist(filter(dht_consensus, FOXA1, GATA3, TFAP2B, H3K27ac)$gene_name),
n_peaks = table(unlist(dht_consensus$gene_name))[gene_name] %>%
as.integer(),
n_peaks = ifelse(is.na(n_peaks), 0, n_peaks)
) %>%
distinct(gene_name, .keep_all = TRUE)
goseq_noar <- structure(
mapped_noar$mapped_to_all, names = mapped_noar$gene_name
) %>%
nullp(bias.data = mapped_noar$n_peaks) %>%
goseq(gene2cat = genes_by_subtype) %>%
as_tibble() %>%
dplyr::select(
subtype = category,
p = over_represented_pvalue,
n_mapped = numDEInCat,
signature_size = numInCat
) %>%
mutate(fdr = p.adjust(p, "fdr"))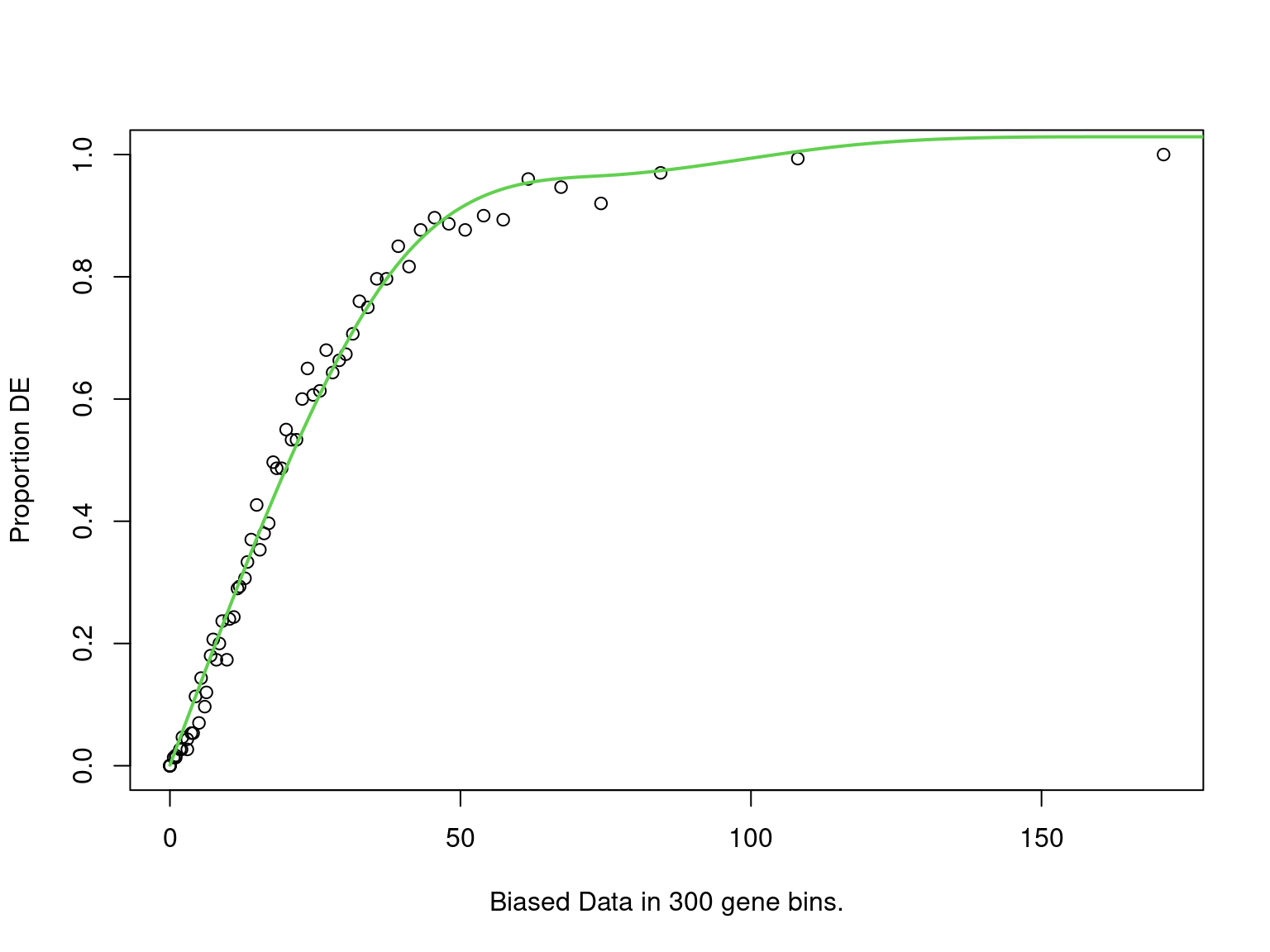
PWF for biased sampling using the number of peaks mapped to a gene as the bias offset
goseq_noar %>%
mutate(`%` = percent(n_mapped / signature_size, accuracy = 0.1)) %>%
dplyr::select(
Subtype = subtype, `Signature Size` = signature_size, `# Mapped` = n_mapped,
`% Mapped` = `%`, p, FDR = fdr
) %>%
pander(
justify = "lrrrrr",
caption = paste(
"Results for Subtype enrichment testing using only genes mapped to a",
"consensus peak *associated with H3K27ac activity*, and where FOXA1",
"GATA3 and TFAP2B were considered to be present, but in the absence of AR."
),
emphasize.strong.rows = which(.$FDR < 0.05)
)| Subtype | Signature Size | # Mapped | % Mapped | p | FDR |
|---|---|---|---|---|---|
| CLCA2 | 136 | 97 | 71.3% | 0.0004744 | 0.00427 |
| ESR1 | 868 | 475 | 54.7% | 0.0964 | 0.4338 |
| ADM | 123 | 64 | 52.0% | 0.2656 | 0.7969 |
| MX1 | 106 | 46 | 43.4% | 0.5812 | 0.9776 |
| FABP4 | 50 | 22 | 44.0% | 0.6161 | 0.9776 |
| TPX2 | 333 | 175 | 52.6% | 0.6816 | 0.9776 |
| DCN | 766 | 387 | 50.5% | 0.7657 | 0.9776 |
| GZMA | 195 | 83 | 42.6% | 0.9199 | 0.9776 |
| CD83 | 400 | 176 | 44.0% | 0.9776 | 0.9776 |
Visualisations: Luminal Genes
MYC
As the gene MYC has been identified as a key target, the binding patterns within the gene were explored. The two binding sites 1) with all four ChIP targets detected, 2) within 100kb of MYC, and 3) which were connected using HiChIP interactions were visualised and exported.
gn <- "MYC"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
arrange(d) %>%
filter(d < 1e5)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) <= 5e3),
hicsize = 3,
max = 5e7,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
cytobands = grch37.cytobands,
zoom = 1.1,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
showAxis = TRUE,
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black",
ylim = c(0,150),
yTicksAt = c(0,150),
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"),
type = "h"
)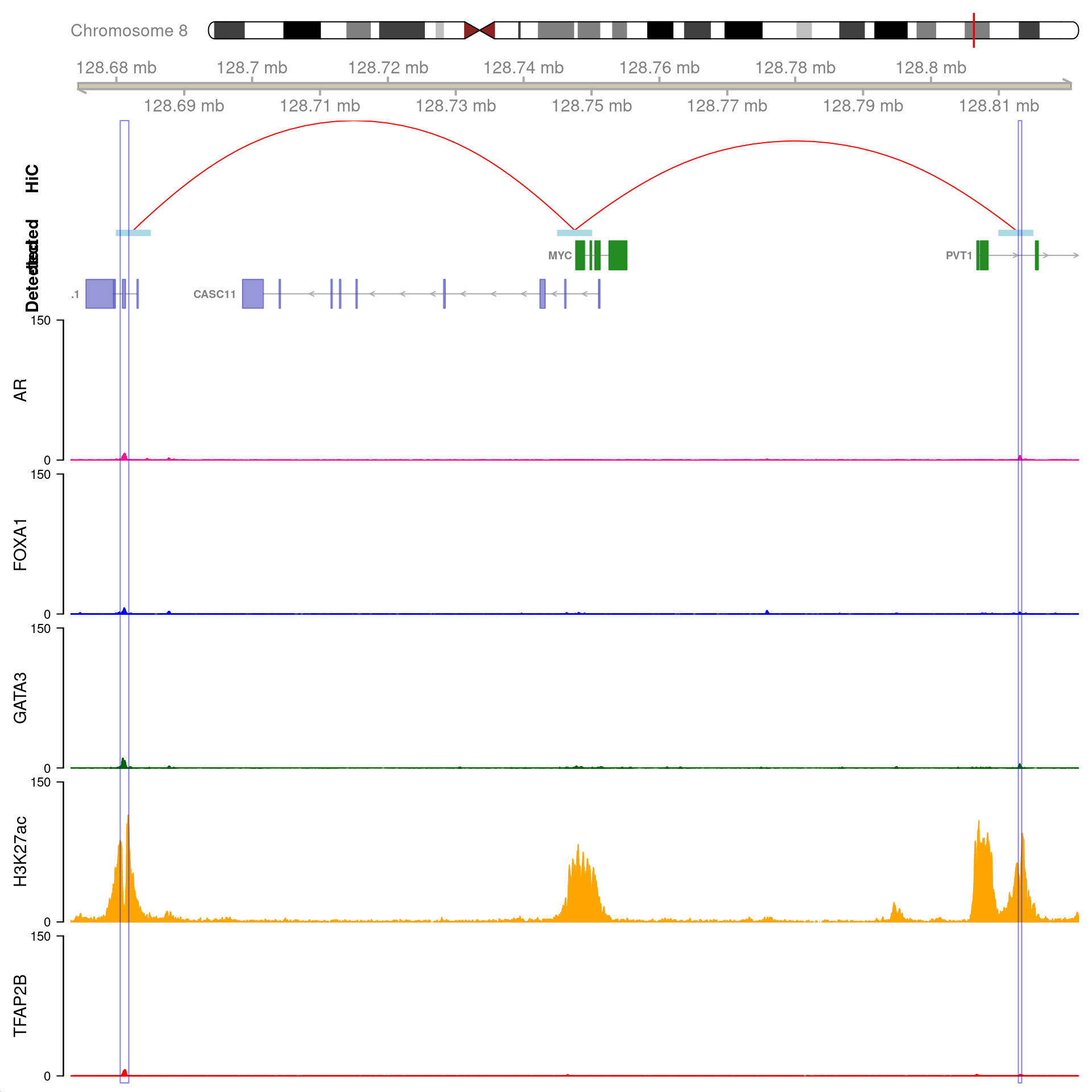
Two joint binding sites either side of MYC connected by HiChIP interactions to the promoter. HiChIP connections were detected at all bin sizes, however, only fine-resolutions 5kb interaction bins are shown.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 3*width(gr), fix = 'center')
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$col <- "grey50"
hl@dp@pars$lwd <- 0.8
hl@dp@pars$inBackground <- FALSE
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()GATA3
gn <- "GATA3"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
arrange(d) %>%
subsetByOverlaps(
hic %>%
subset(as.integer(bin_size) == 5e3) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
)
)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) <= 5e3),
hicsize = 3,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
cytobands = grch37.cytobands,
zoom = 1.1,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
showAxis = TRUE,
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black",
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150) #to avoid the labels clashing
)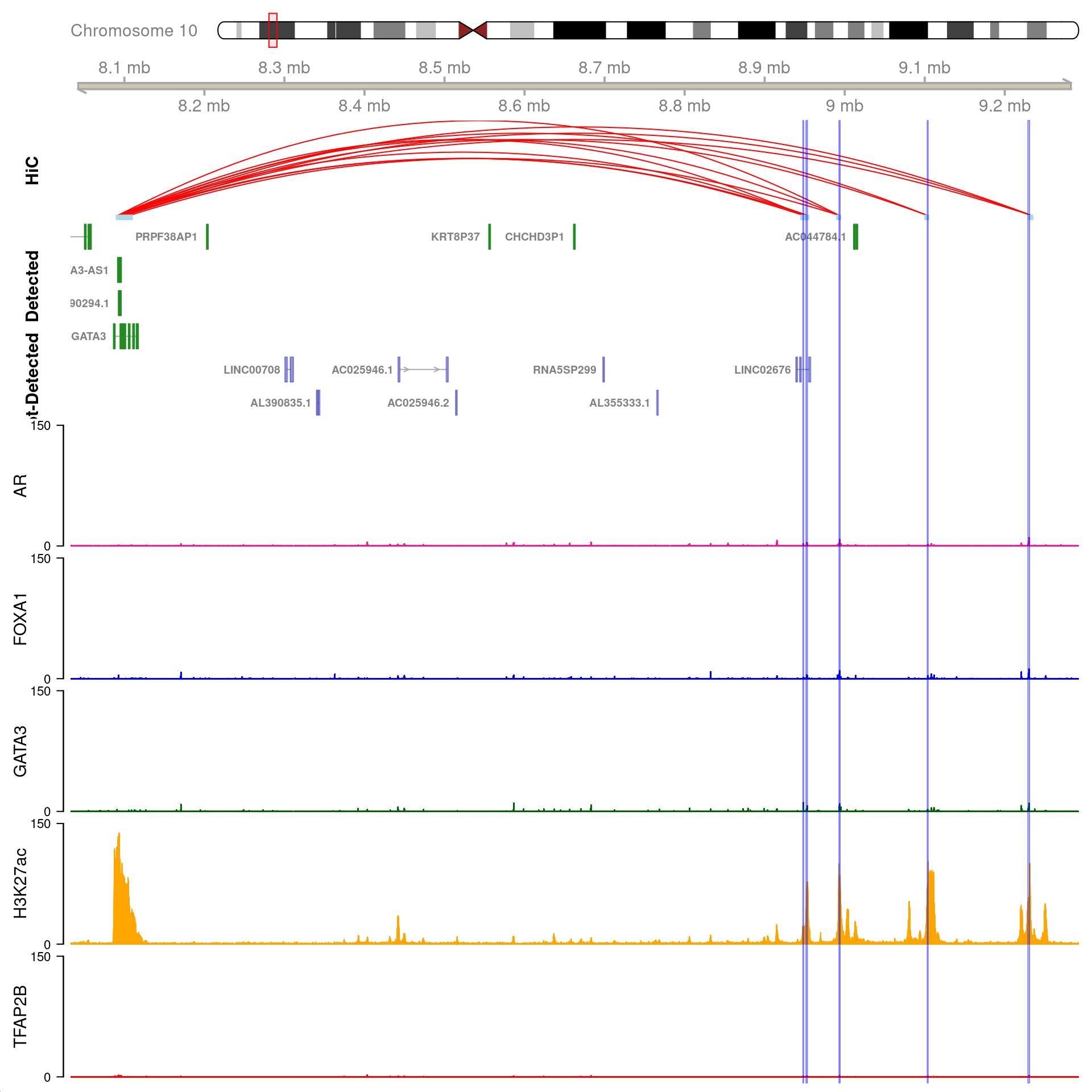
All H3K27ac-associated peaks where all four targets were detected, and which map to GATA3. Only the high-resolution 5kb bins are shown.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
region_plot$Detected@dp@pars$just.group <- "right"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 3*width(gr), fix = 'center')
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$col <- "grey50"
hl@dp@pars$lwd <- 0.8
hl@dp@pars$inBackground <- FALSE #puts the blue box in front of the other dat
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()FOXA1
gn <- "FOXA1"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
arrange(d) %>%
subsetByOverlaps(
hic %>%
subset(as.integer(bin_size) == 5e3) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
)
)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) <= 5e3),
max = 1e5,
hicsize = 3,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
cytobands = grch37.cytobands,
zoom = 1.2,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black",
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150), #to avoid the labels clashing
showAxis = TRUE, #to plot read number
)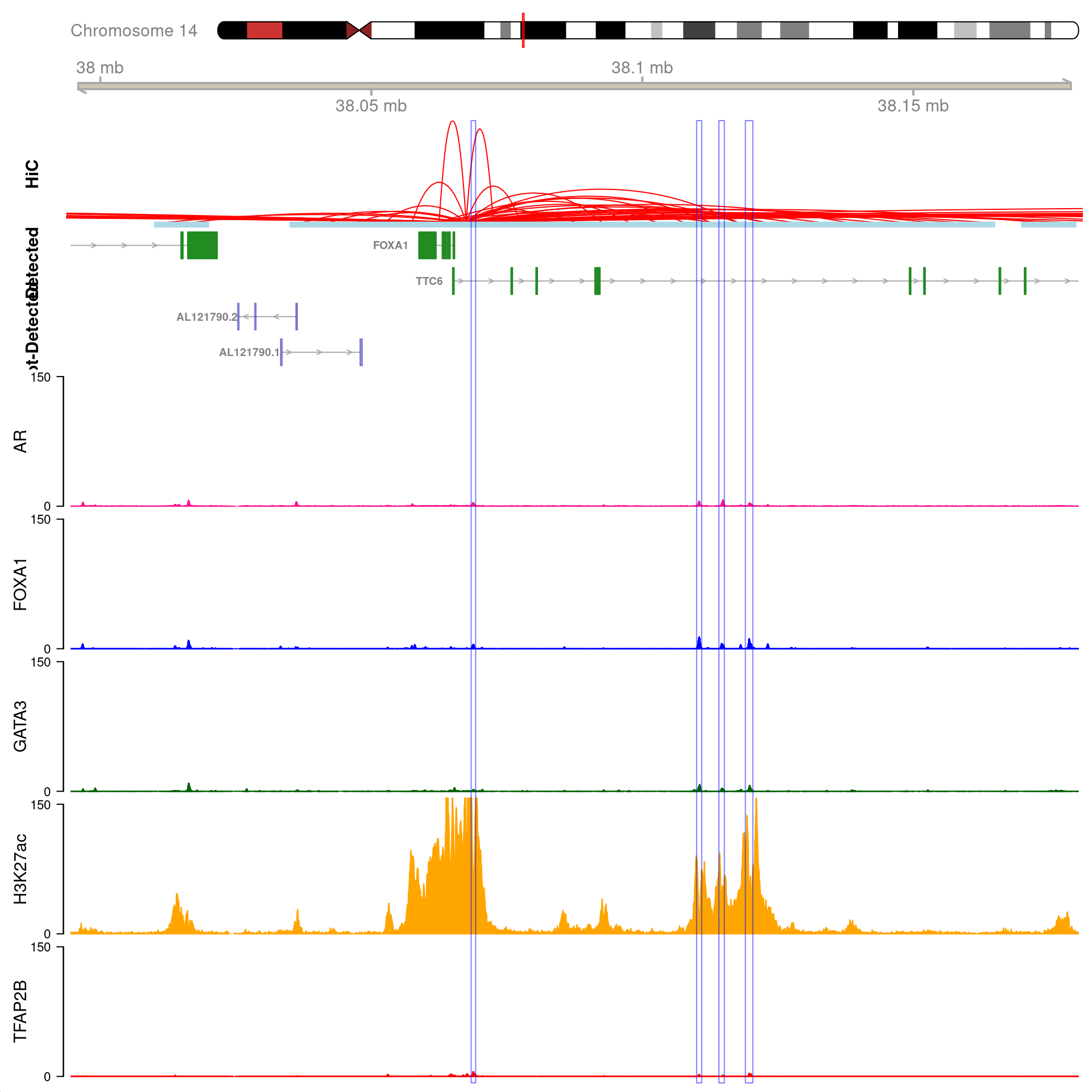
All H3K27ac-associated peaks where all four targets were detected, and which map to FOXA1. Only the high-resolution 5kb bins are shown.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 1.5 * width(gr), fix = 'center')
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$col <- "grey50" #blue clashes with the FOXA1 track
hl@dp@pars$inBackground <- FALSE #puts the grey box in front of the other dat
hl@dp@pars$lwd <- 0.8
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()XBP1
gn <- "XBP1"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
arrange(d) %>%
filter(d < 100e3)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) == 5e3),
hicsize = 3,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150), #to avoid the labels clashing
showAxis = TRUE, #to plot read number
cytobands = grch37.cytobands,
zoom = 1.1,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black"
)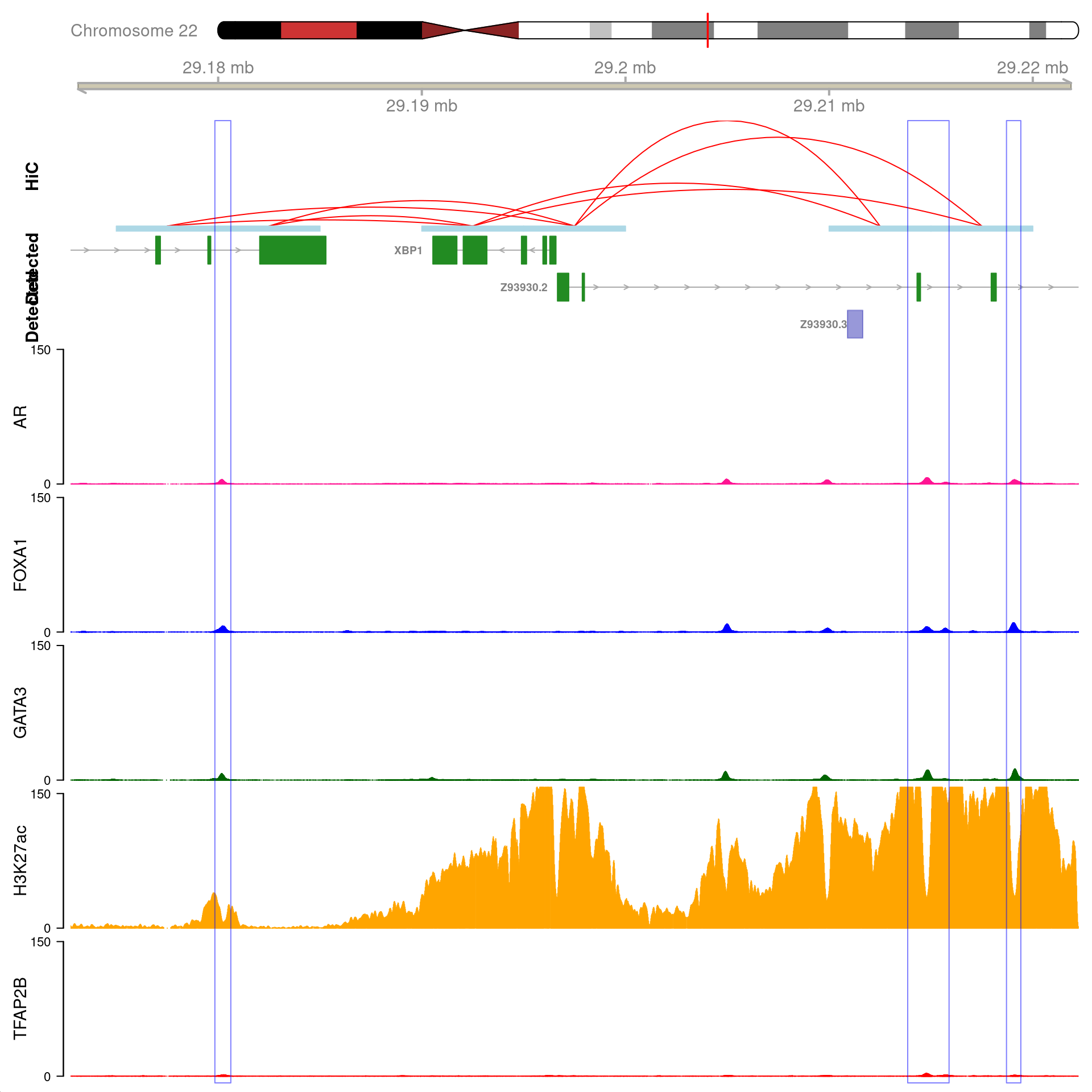
All H3K27ac-associated peaks where all four targets were detected, and which are within 100kb of XBP1. Only the high-resolution 5kb interaction bins are shown. The two additional peaks were not considered to show evidence of TFAP2B binding by macs2 callpeak and are not highlighted for this reason
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 1.5 * width(gr), fix = 'center')
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$inBackground <- FALSE #puts the grey box in front of the other dat
hl@dp@pars$col <- "grey50" #blue clashes with the FOXA1 track
hl@dp@pars$lwd <- 0.8
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()MYB
gn <- "MYB"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
arrange(d)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) == 20e3),
hicsize = 3,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150), #to avoid the labels clashing
showAxis = TRUE, #to plot read number
cytobands = grch37.cytobands,
zoom = 1.1,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black"
)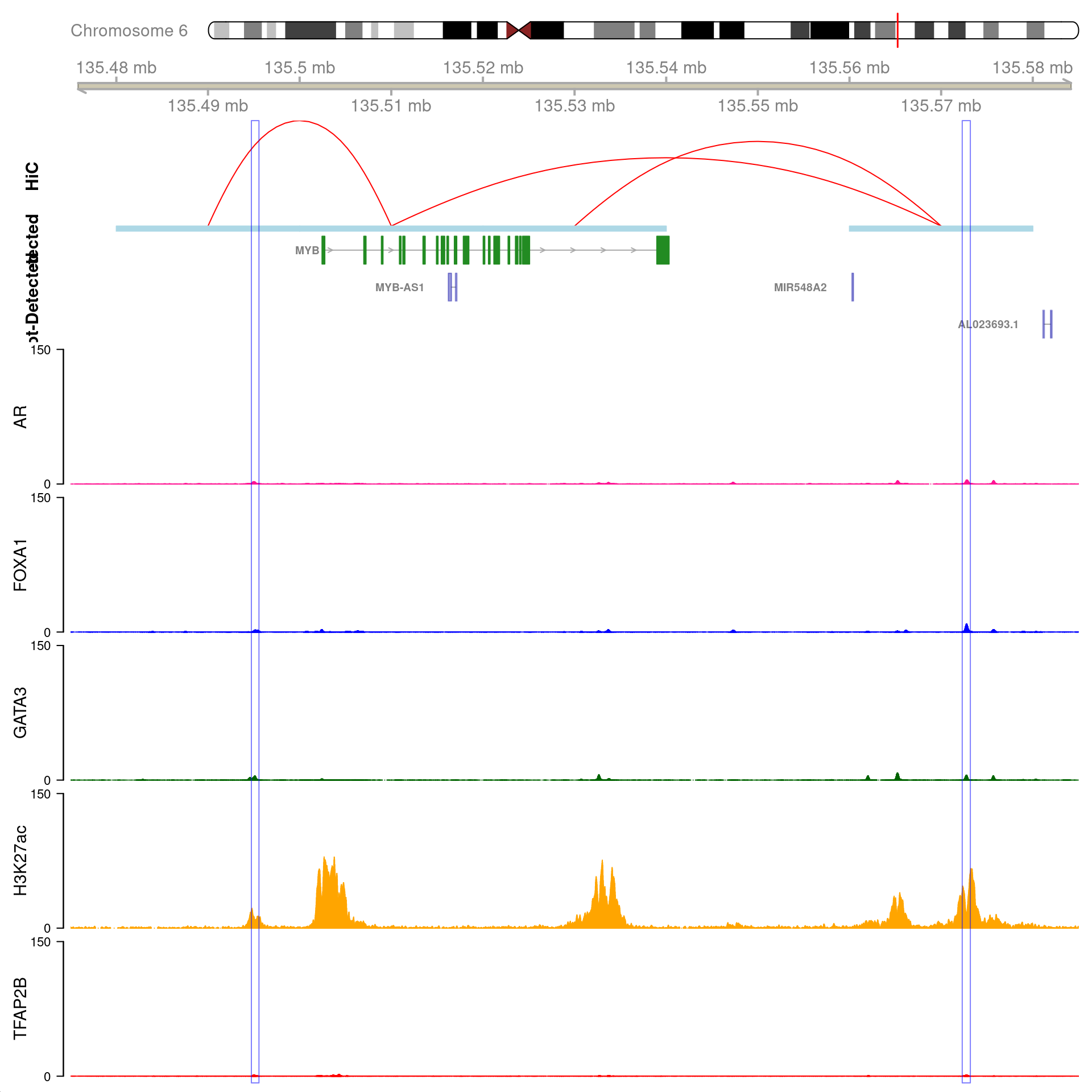
All H3K27ac-associated peaks where all four targets were detected, and which map to MYB. Only the low-resolution 20kb interaction bins are shown.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 1.5 * width(gr), fix = 'center')
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$inBackground <- FALSE #puts the grey box in front of the other dat
hl@dp@pars$col <- "grey50" #blue clashes with the FOXA1 track
hl@dp@pars$lwd <- 0.8
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()AR
ogn <- "AR"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
filter(d < 1e5)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) == 20e3),
hicsize = 3,
max = 5e4,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150), #to avoid the labels clashing
showAxis = TRUE, #to plot read number
cytobands = grch37.cytobands,
zoom = 6,
shift = -6e4,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black"
)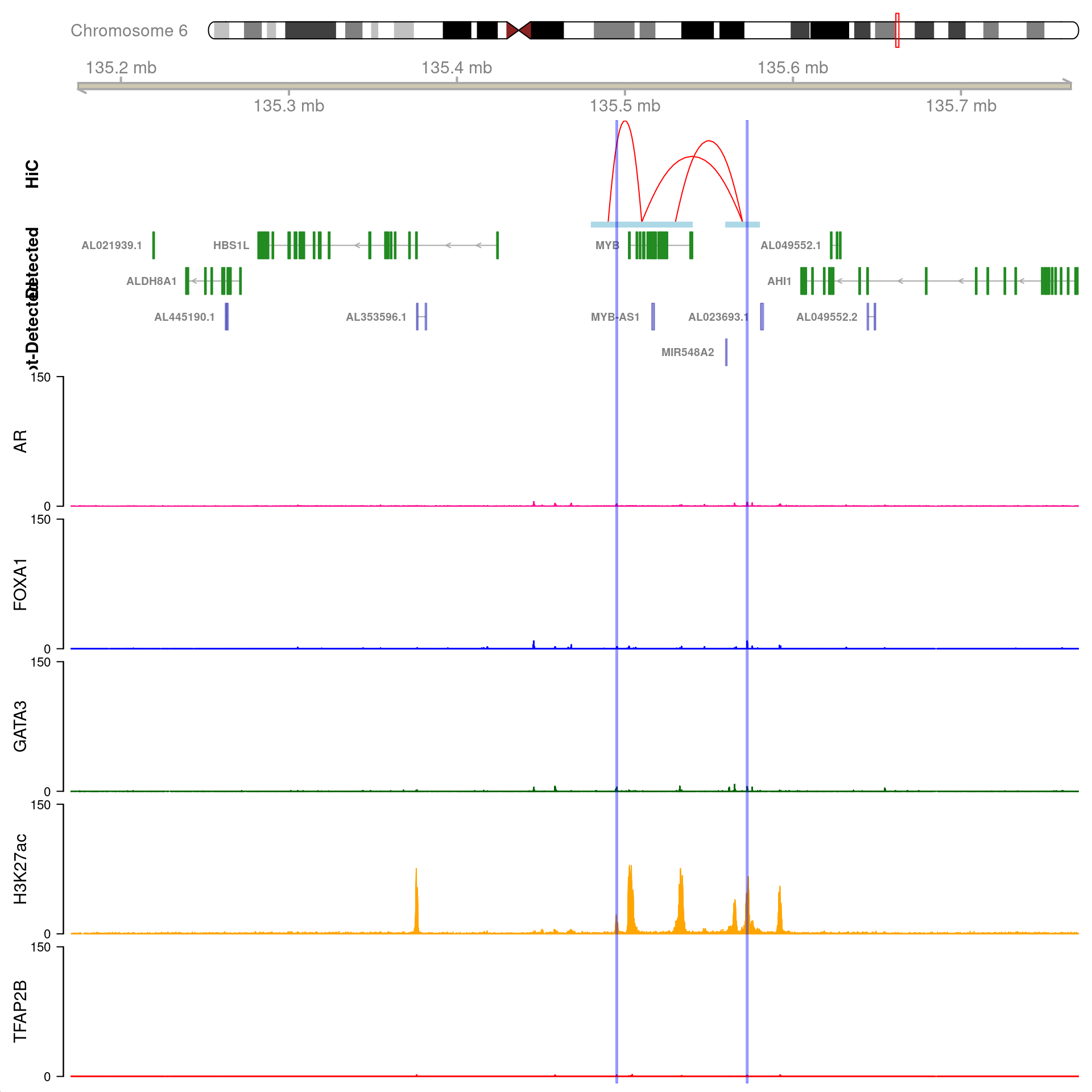
All H3K27ac-associated peaks where all four targets were detected, which map to AR and are within 100kb of the gene. Only the low-resolution 20kb HiC interaction bins are shown.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 3 * width(gr), fix = 'center') %>%
range()
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$inBackground <- FALSE #puts the grey box in front of the other dat
hl@dp@pars$col <- "grey50" #blue clashes with the FOXA1 track
hl@dp@pars$lwd <- 0.8
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()Visualisations:: Apocrine Genes
CLCA2
gn <- "CLCA2"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
arrange(d)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
collapseTranscripts = "meta",
coverage = bwfl,
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150), #to avoid the labels clashing
showAxis = TRUE, #to plot read number
cytobands = grch37.cytobands,
zoom = 4,
shift = 1e4,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black"
)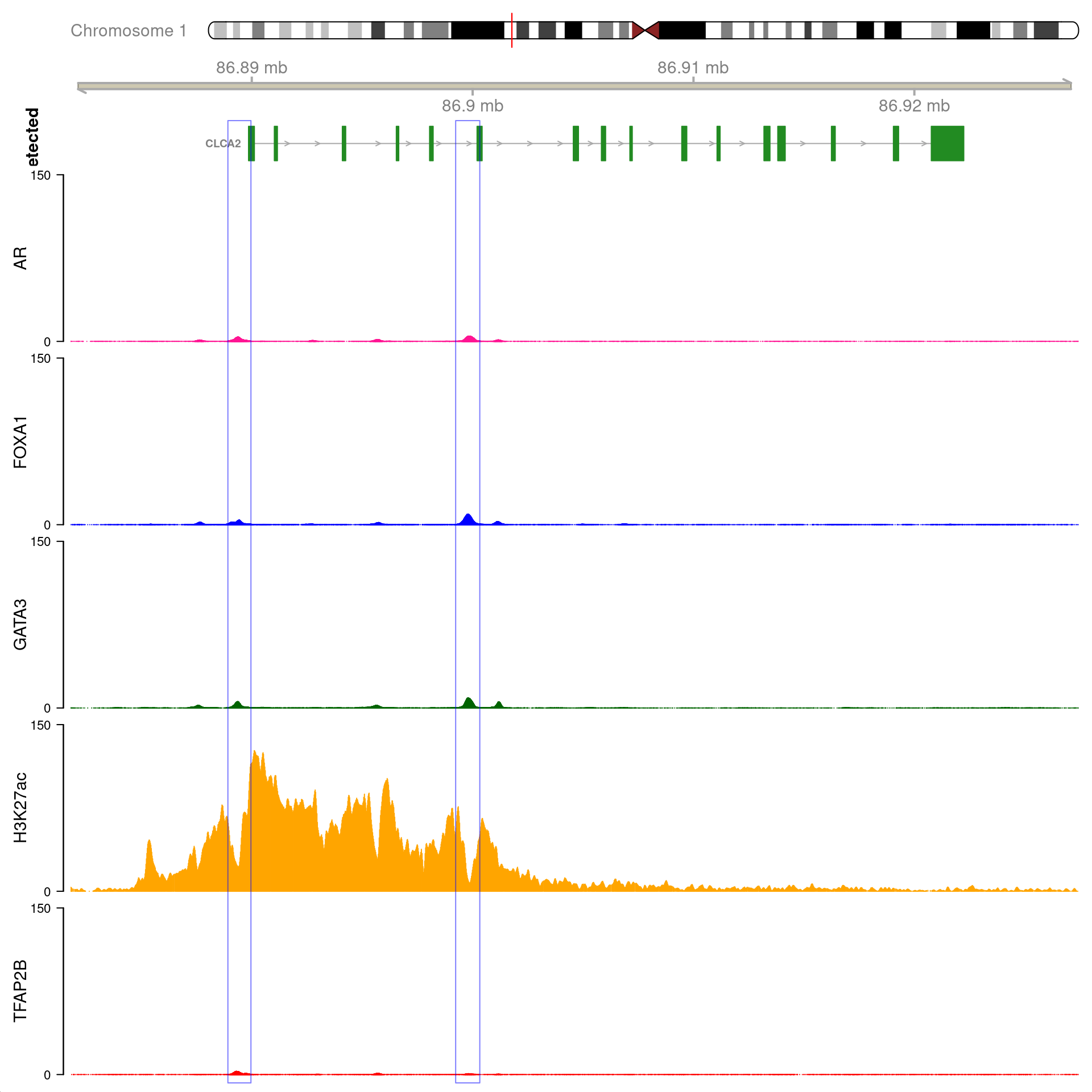
All H3K27ac-associated peaks where all four targets were detected, and which within the transcribed region for CLCA2. No HiC data is shown.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 1.2 * width(gr), fix = 'center')
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$inBackground <- FALSE #puts the grey box in front of the other dat
hl@dp@pars$col <- "grey50" #blue clashes with the FOXA1 track
hl@dp@pars$lwd <- 0.8
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()TFAP2B
gn <- "TFAP2B"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
filter(d < 5e5)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) == 10e3),
hicsize = 3,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150), #to avoid the labels clashing
showAxis = TRUE, #to plot read number
cytobands = grch37.cytobands,
zoom = 1.1,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black"
)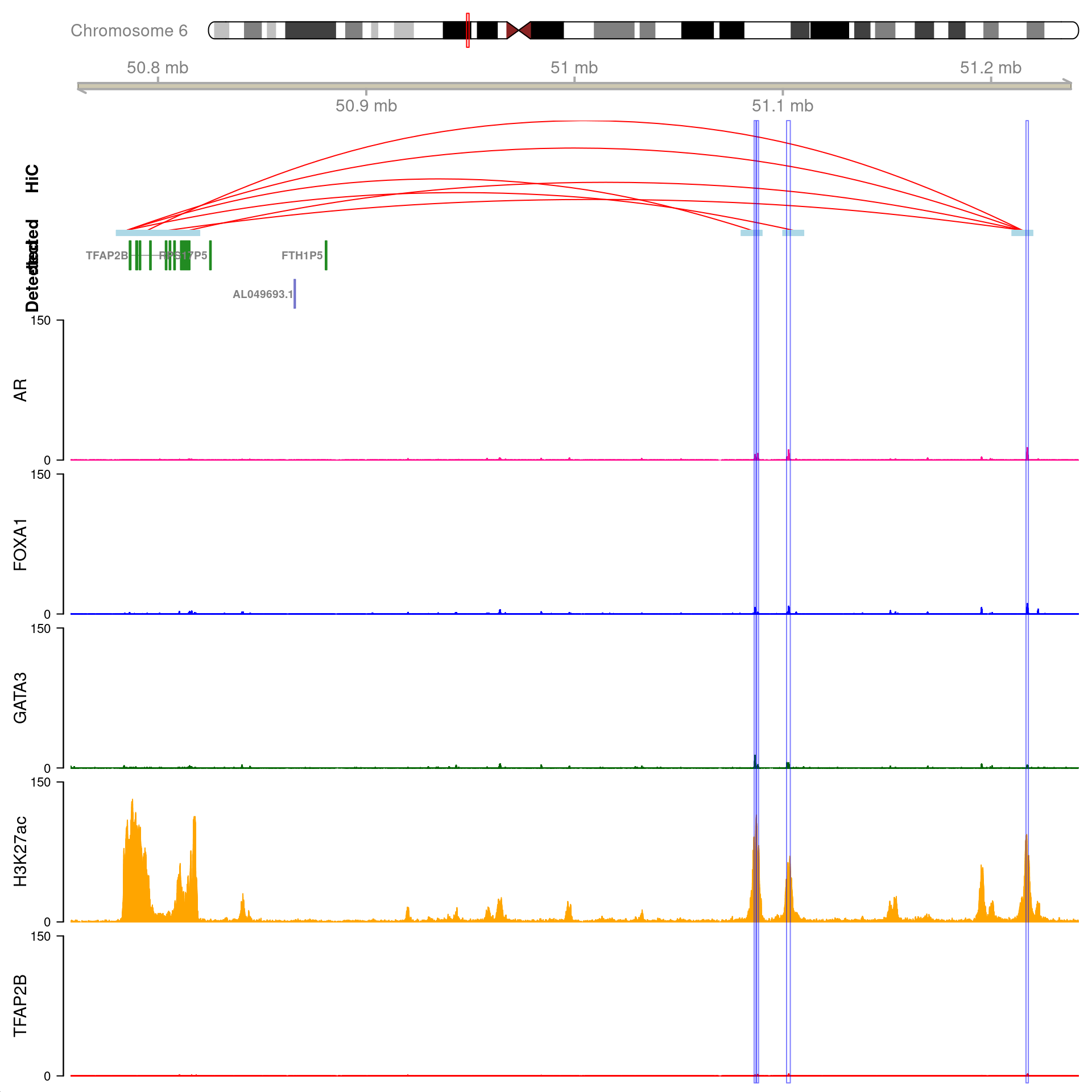
All H3K27ac-associated peaks where all four targets were detected, which map to TFAP2B and are within 500kb of TFAP2B. Only the medium-resolution 10kb interaction bins are shown.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 3 * width(gr), fix = 'center') %>%
reduce()
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$inBackground <- FALSE #puts the grey box in front of the other dat
hl@dp@pars$col <- "grey50" #blue clashes with the FOXA1 track
hl@dp@pars$lwd <- 0.8
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()KYNU
gn <- "KYNU"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn)))
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) <= 40e3),
hicsize = 3,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150), #to avoid the labels clashing
showAxis = TRUE, #to plot read number
cytobands = grch37.cytobands,
zoom = 5,
shift = 3e4,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black"
)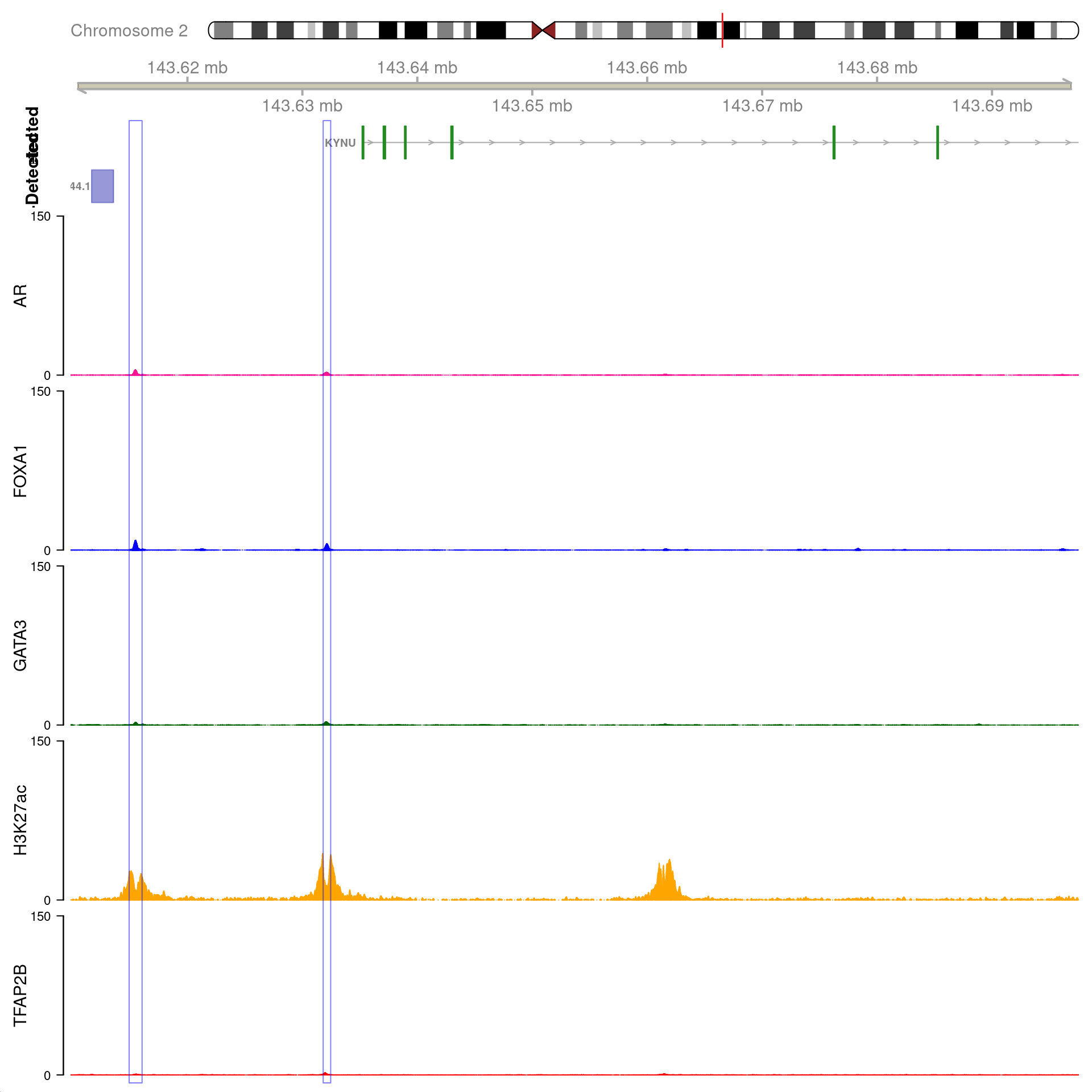
All H3K27ac-associated peaks where all four targets were detected, and which map to KYNU. No HiC interaction bins were found in this region.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 1.5 * width(gr), fix = 'center')
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$inBackground <- FALSE #puts the grey box in front of the other dat
hl@dp@pars$col <- "grey50" #blue clashes with the FOXA1 track
hl@dp@pars$lwd <- 0.8
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.1*width(.), fix = 'center') %>%
resize(width = 0.4 * width(.), fix = 'start')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()FGFR4
gn <- "FGFR4"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
filter(d < 1e5)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) <= 5e3),
hicsize = 3,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150), #to avoid the labels clashing
showAxis = TRUE, #to plot read number
cytobands = grch37.cytobands,
zoom = 1.1,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black"
)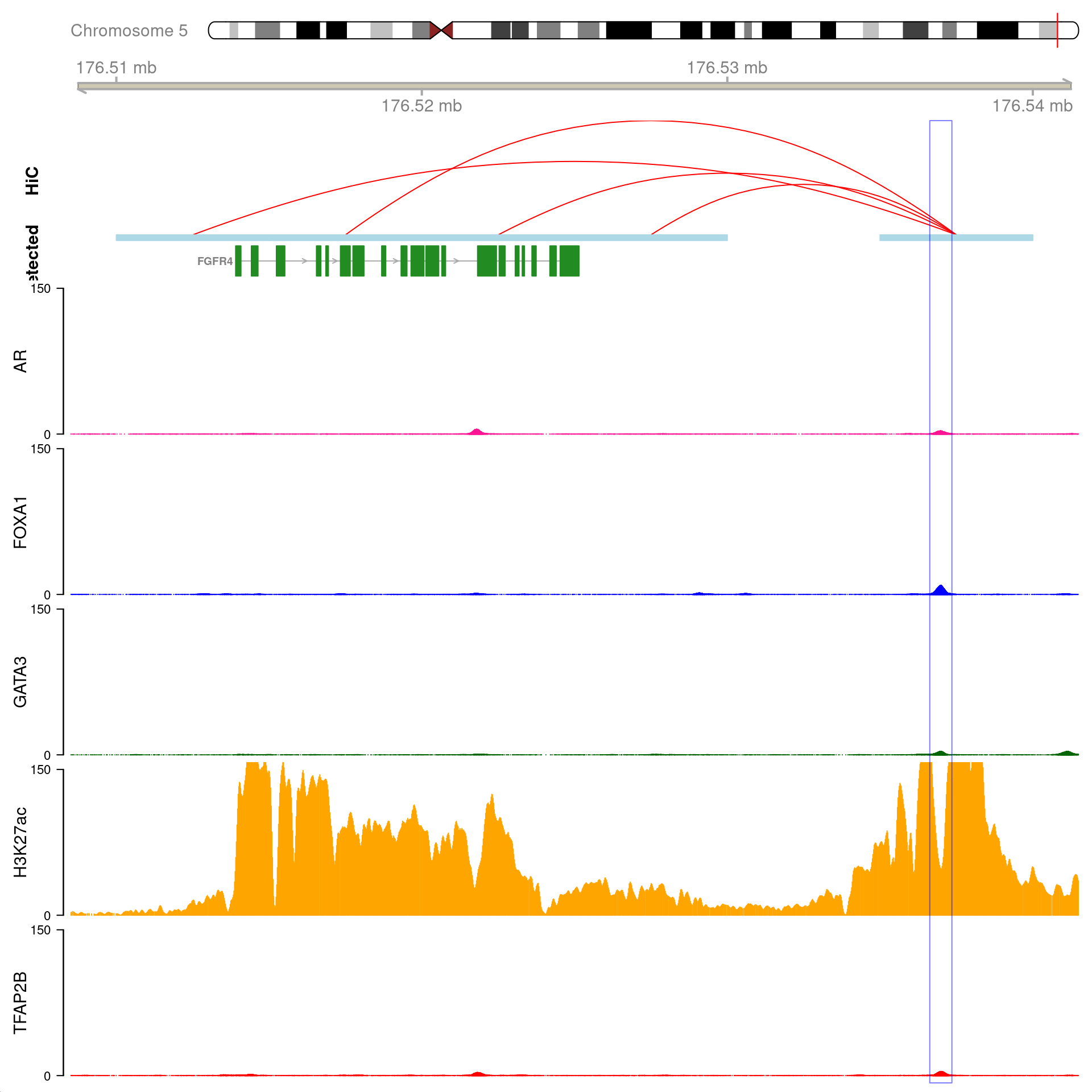
All H3K27ac-associated peaks where all four targets were detected, which map to FGFR4 and are within 100kb of the gene. Only the high-resolution 5kb HiC interaction bins are shown.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 2 * width(gr), fix = 'center')
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$inBackground <- FALSE #puts the grey box in front of the other dat
hl@dp@pars$col <- "grey50" #blue clashes with the FOXA1 track
hl@dp@pars$lwd <- 0.8
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()PGAP3
gn <- "PGAP3"
gr <- dht_consensus %>%
filter(!!!syms(targets), mapped, H3K27ac) %>%
filter(
vapply(gene_name, function(x) gn %in% x, logical(1))
) %>%
mutate(d = distance(., subset(all_gr$gene, gene_name == gn))) %>%
filter(d < 1e5)
fs <- 14
region_plot <- plotHFGC(
gr = gr,
hic = hic %>%
subsetByOverlaps(gr) %>%
subsetByOverlaps(
subset(all_gr$gene, gene_name == gn)
) %>%
subset(as.integer(bin_size) <= 5e3),
hicsize = 3,
max = 5e4,
genes = tm %>%
mutate(detected = ifelse(gene %in% detected, "Detected", "Not-Detected")) %>%
split(.$detected),
genecol = c(
Detected = "forestgreen", "Not-Detected" = rgb(0.2, 0.2, 0.7, 0.5)
),
coverage = bwfl,
linecol = c("deeppink", "blue", "darkgreen", "orange", "red"), #can choose any colours
type = "h", #plot histograms
ylim = c(0,150), #compromise
yTicksAt = c(0,150), #to avoid the labels clashing
showAxis = TRUE, #to plot read number
cytobands = grch37.cytobands,
zoom = 1.1,
highlight = rgb(0, 0, 1, 0.5),
title.width = 1,
col.title = "black", background.title = "white",
rotation.title = 90, fontsize = fs,
fontface.title = 1,
legend = FALSE,
fontcolor.legend = "black"
)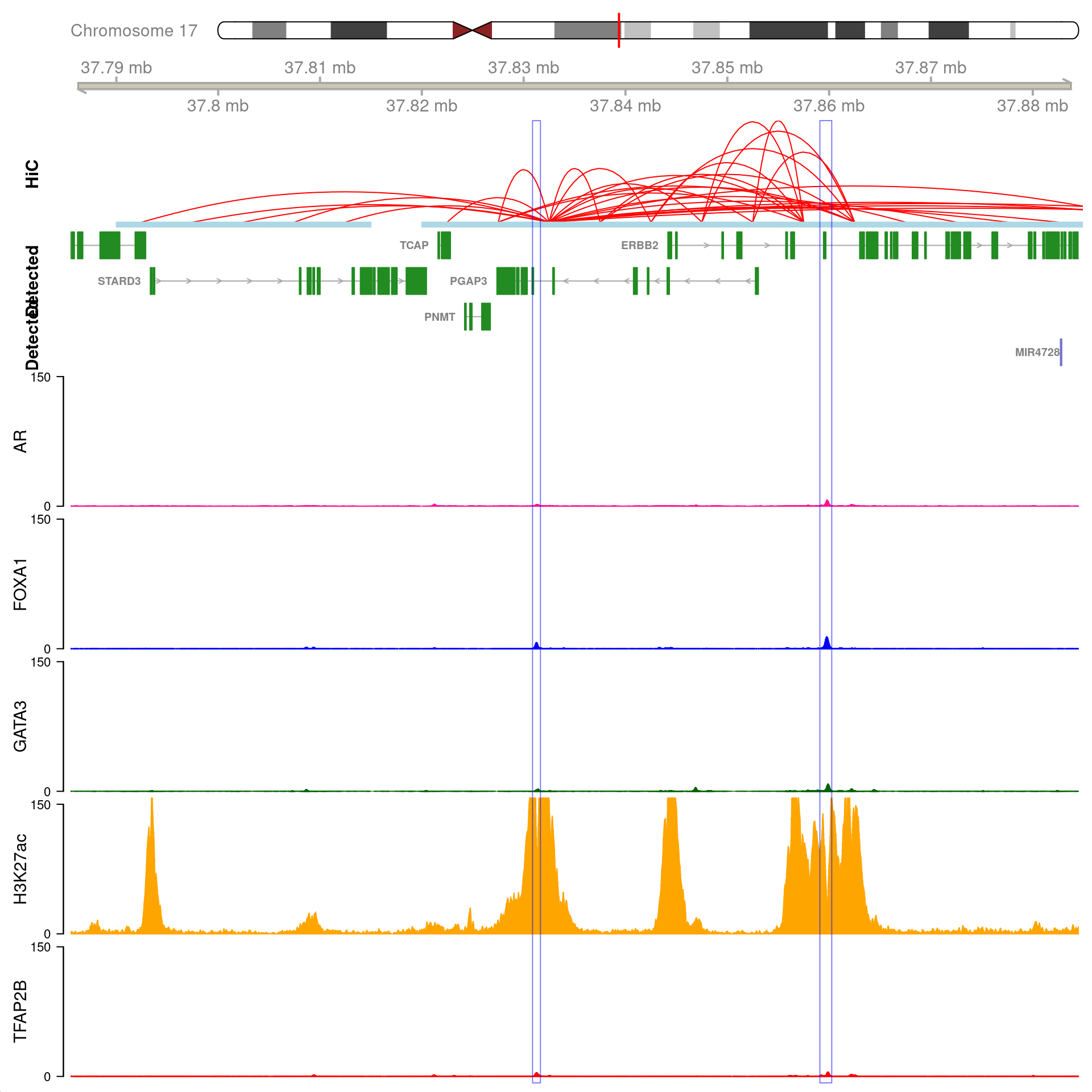
All H3K27ac-associated peaks where all four targets were detected, which map to PGAP3 and are within 100kb of the gene. Only the high-resolution 5kb HiC interaction bins are shown.
region_plot[[1]]@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$fontcolor <- "black"
region_plot$Axis@dp@pars$col <- "grey30"
region_plot$AR@dp@pars$ylim <- c(0,max(region_plot$AR@data + 1))
region_plot$AR@dp@pars$yTicksAt = c(0,round(max(region_plot$AR@data),0))
region_plot$TFAP2B@dp@pars$ylim <- c(0,max(region_plot$TFAP2B@data + 1))
region_plot$TFAP2B@dp@pars$yTicksAt = c(0,round(max(region_plot$TFAP2B@data),0))
region_plot$GATA3@dp@pars$ylim <- c(0,max(region_plot$GATA3@data + 1))
region_plot$GATA3@dp@pars$yTicksAt = c(0,round(max(region_plot$GATA3@data),0))
region_plot$FOXA1@dp@pars$ylim <- c(0,max(region_plot$FOXA1@data + 1))
region_plot$FOXA1@dp@pars$yTicksAt = c(0,round(max(region_plot$FOXA1@data),0))
region_plot$H3K27ac@dp@pars$ylim <- c(0,max(region_plot$H3K27ac@data + 1))
region_plot$H3K27ac@dp@pars$yTicksAt = c(0,round(max(region_plot$H3K27ac@data),0))
if ("HiC" %in% names(region_plot)) {
region_plot$HiC@name <- "HiChIP"
region_plot$HiC@dp@pars$fontface.title <- 1
region_plot$HiC@dp@pars$rotation.title <- 0
}
region_plot$Detected@dp@pars$fontface.title <- 1
region_plot$Detected@dp@pars$rotation.title <- 0
region_plot$Detected@dp@pars$fontsize.group <- fs
region_plot$Detected@dp@pars$cex.group <- 0.8
region_plot$Detected@dp@pars$fontface.group <- 1
region_plot$Detected@dp@pars$fontcolor.group <- "black"
if ("Not-Detected" %in% names(region_plot)) {
region_plot$`Not-Detected`@dp@pars$fontcolor.group <- "black"
region_plot$`Not-Detected`@dp@pars$fontface.title <- 1
region_plot$`Not-Detected`@dp@pars$rotation.title <- 0
region_plot$`Not-Detected`@dp@pars$fontsize.group <- fs
region_plot$`Not-Detected`@dp@pars$cex.group <- 0.8
region_plot$`Not-Detected`@dp@pars$fontface.group <- 1
region_plot$`Not-Detected`@dp@pars$just.group <- "right"
}
region_plot$AR@dp@pars$rotation.title <- 0
region_plot$FOXA1@dp@pars$rotation.title <- 0
region_plot$GATA3@dp@pars$rotation.title <- 0
region_plot$TFAP2B@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$rotation.title <- 0
region_plot$H3K27ac@dp@pars$size <- 5
ax <- region_plot[1:2]
top_panels <- region_plot[names(region_plot) %in% c("HiC", "Detected", "Not-Detected")]
hl <- HighlightTrack(
region_plot[c(targets, "H3K27ac")],
range = resize(gr, width = 1.5 * width(gr), fix = 'center')
)
hl@dp@pars$fill <- rgb(1, 1, 1, 0)
hl@dp@pars$inBackground <- FALSE #puts the grey box in front of the other dat
hl@dp@pars$col <- "grey50" #blue clashes with the FOXA1 track
hl@dp@pars$lwd <- 0.8
pdf(here::here("output", glue("{gn}.pdf")), width = 10, height = 10)
plot_range <- c(gr, subset(all_gr$gene, gene_name == gn)) %>%
range(ignore.strand = TRUE) %>%
resize(width = 1.2*width(.), fix = 'center')
Gviz::plotTracks(
c(ax, top_panels, hl),
from = start(plot_range), to = end(plot_range),
title.width = 1,
margin = 40,
fontface.main = 1
)
dev.off()Data Export
Sequence Export
hg19 <- BSgenome.Hsapiens.UCSC.hg19
sq19 <- seqinfo(hg19)
consenus_hg19 <- dht_consensus
seqinfo(consenus_hg19) <- sq19
features_hg19 <- features
seqinfo(features_hg19) <- sq19
getSeq(
hg19,
consenus_hg19 %>%
filter(AR, FOXA1, GATA3, !TFAP2B) %>%
resize(width = 500, fix = 'center') %>%
setNames(as.character(.))
) %>%
writeXStringSet(
here::here("output", "ar_foxa1_gata3_no_tfap2b.fa")
)
getSeq(
hg19,
consenus_hg19 %>%
filter(AR, FOXA1, GATA3, TFAP2B) %>%
resize(width = 500, fix = 'center') %>%
setNames(as.character(.))
) %>%
writeXStringSet(
here::here("output", "ar_foxa1_gata3_tfap2b.fa")
)
getSeq(
hg19,
consenus_hg19 %>%
filter(AR, FOXA1, GATA3, TFAP2B, H3K27ac) %>%
resize(width = 500, fix = 'center') %>%
setNames(as.character(.))
) %>%
writeXStringSet(
here::here("output", "all4_h3k27ac.fa")
)
getSeq(
hg19,
consenus_hg19 %>%
filter(AR, FOXA1, GATA3, TFAP2B, !H3K27ac) %>%
resize(width = 500, fix = 'center') %>%
setNames(as.character(.))
) %>%
writeXStringSet(
here::here("output", "all4_no_h3k27ac.fa")
)Peaks
write_rds(dht_consensus, here::here("output/dht_consensus.rds"), compress = "gz")
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] BSgenome.Hsapiens.UCSC.hg19_1.4.3 BSgenome_1.64.0
[3] Biostrings_2.64.0 XVector_0.36.0
[5] scico_1.3.0 cowplot_1.1.1
[7] goseq_1.48.0 geneLenDataBase_1.32.0
[9] BiasedUrn_1.07 corrplot_0.92
[11] readxl_1.4.0 Gviz_1.40.1
[13] effects_4.2-1 carData_3.0-5
[15] nnet_7.3-17 glue_1.6.2
[17] multcomp_1.4-19 TH.data_1.1-1
[19] MASS_7.3-56 survival_3.2-13
[21] mvtnorm_1.1-3 GenomicInteractions_1.30.0
[23] InteractionSet_1.24.0 rtracklayer_1.56.0
[25] UpSetR_1.4.0 htmltools_0.5.2
[27] reactable_0.3.0 scales_1.2.0
[29] pander_0.6.5 plyranges_1.16.0
[31] extraChIPs_1.0.1 SummarizedExperiment_1.26.1
[33] Biobase_2.56.0 MatrixGenerics_1.8.0
[35] matrixStats_0.62.0 GenomicRanges_1.48.0
[37] GenomeInfoDb_1.32.2 IRanges_2.30.0
[39] S4Vectors_0.34.0 BiocGenerics_0.42.0
[41] BiocParallel_1.30.2 magrittr_2.0.3
[43] forcats_0.5.1 stringr_1.4.0
[45] dplyr_1.0.9 purrr_0.3.4
[47] readr_2.1.2 tidyr_1.2.0
[49] tibble_3.1.7 ggplot2_3.3.6
[51] tidyverse_1.3.1 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] rappdirs_0.3.3 csaw_1.30.1 bit64_4.0.5
[4] knitr_1.39 DelayedArray_0.22.0 data.table_1.14.2
[7] rpart_4.1.16 KEGGREST_1.36.0 RCurl_1.98-1.6
[10] AnnotationFilter_1.20.0 doParallel_1.0.17 generics_0.1.2
[13] GenomicFeatures_1.48.3 callr_3.7.0 EnrichedHeatmap_1.26.0
[16] RSQLite_2.2.14 bit_4.0.4 tzdb_0.3.0
[19] xml2_1.3.3 lubridate_1.8.0 httpuv_1.6.5
[22] assertthat_0.2.1 xfun_0.31 hms_1.1.1
[25] jquerylib_0.1.4 evaluate_0.15 promises_1.2.0.1
[28] fansi_1.0.3 restfulr_0.0.13 progress_1.2.2
[31] dbplyr_2.1.1 igraph_1.3.1 DBI_1.1.2
[34] htmlwidgets_1.5.4 ellipsis_0.3.2 crosstalk_1.2.0
[37] backports_1.4.1 insight_0.17.1 survey_4.1-1
[40] biomaRt_2.52.0 vctrs_0.4.1 here_1.0.1
[43] ensembldb_2.20.1 cachem_1.0.6 withr_2.5.0
[46] ggforce_0.3.3 vroom_1.5.7 checkmate_2.1.0
[49] GenomicAlignments_1.32.0 prettyunits_1.1.1 cluster_2.1.3
[52] lazyeval_0.2.2 crayon_1.5.1 labeling_0.4.2
[55] edgeR_3.38.1 pkgconfig_2.0.3 tweenr_1.0.2
[58] nlme_3.1-157 ProtGenerics_1.28.0 rlang_1.0.2
[61] lifecycle_1.0.1 sandwich_3.0-1 filelock_1.0.2
[64] BiocFileCache_2.4.0 modelr_0.1.8 dichromat_2.0-0.1
[67] cellranger_1.1.0 rprojroot_2.0.3 polyclip_1.10-0
[70] reactR_0.4.4 Matrix_1.4-1 boot_1.3-28
[73] zoo_1.8-10 reprex_2.0.1 base64enc_0.1-3
[76] whisker_0.4 GlobalOptions_0.1.2 processx_3.5.3
[79] viridisLite_0.4.0 png_0.1-7 rjson_0.2.21
[82] bitops_1.0-7 getPass_0.2-2 blob_1.2.3
[85] shape_1.4.6 jpeg_0.1-9 memoise_2.0.1
[88] plyr_1.8.7 zlibbioc_1.42.0 compiler_4.2.0
[91] scatterpie_0.1.7 BiocIO_1.6.0 RColorBrewer_1.1-3
[94] clue_0.3-61 lme4_1.1-29 Rsamtools_2.12.0
[97] cli_3.3.0 ps_1.7.0 htmlTable_2.4.0
[100] Formula_1.2-4 mgcv_1.8-40 ggside_0.2.0.9990
[103] tidyselect_1.1.2 stringi_1.7.6 highr_0.9
[106] mitools_2.4 yaml_2.3.5 locfit_1.5-9.5
[109] latticeExtra_0.6-29 ggrepel_0.9.1 sass_0.4.1
[112] VariantAnnotation_1.42.1 tools_4.2.0 parallel_4.2.0
[115] circlize_0.4.15 rstudioapi_0.13 foreach_1.5.2
[118] foreign_0.8-82 git2r_0.30.1 metapod_1.4.0
[121] gridExtra_2.3 farver_2.1.0 digest_0.6.29
[124] Rcpp_1.0.8.3 broom_0.8.0 later_1.3.0
[127] httr_1.4.3 AnnotationDbi_1.58.0 biovizBase_1.44.0
[130] ComplexHeatmap_2.12.0 colorspace_2.0-3 rvest_1.0.2
[133] XML_3.99-0.9 fs_1.5.2 splines_4.2.0
[136] jsonlite_1.8.0 nloptr_2.0.3 ggfun_0.0.6
[139] R6_2.5.1 Hmisc_4.7-0 pillar_1.7.0
[142] fastmap_1.1.0 minqa_1.2.4 codetools_0.2-18
[145] utf8_1.2.2 lattice_0.20-45 bslib_0.3.1
[148] curl_4.3.2 GO.db_3.15.0 limma_3.52.1
[151] rmarkdown_2.14 munsell_0.5.0 GetoptLong_1.0.5
[154] GenomeInfoDbData_1.2.8 iterators_1.0.14 haven_2.5.0
[157] gtable_0.3.0