Enrichment Analysis
Stephen Pederson
Dame Roma Mitchell Cancer Research Laboratories
Adelaide Medical School
University of Adelaide
03 November, 2020
Last updated: 2020-11-03
Checks: 7 0
Knit directory: MFM-223_DHT-RNASeq/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200930) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version cbf7fbd. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Unstaged changes:
Modified: analysis/_site.yml
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/dge_enrichment.Rmd) and HTML (docs/dge_enrichment.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | cbf7fbd | Steve Ped | 2020-11-03 | Rebuilt all after reorganising Rmd files |
| html | bcdc500 | Steve Ped | 2020-10-15 | Build site. |
| Rmd | 2f025f7 | Steve Pederson | 2020-10-14 | Reset for run with correct sjdbOverhang |
library(tidyverse)
library(yaml)
library(scales)
library(pander)
library(glue)
library(edgeR)
library(cowplot)
library(magrittr)
library(ggrepel)
library(DT)
library(msigdbr)
library(goseq)
library(reactable)
library(htmltools)
library(tidygraph)
library(ggraph)
library(corrplot)panderOptions("table.split.table", Inf)
panderOptions("big.mark", ",")
panderOptions("missing", "")
theme_set(theme_bw())as_sci <- function(p, d = 2, min = 0.01){
fmt <- glue("%.{d}e")
new <- character(length(p))
new[p > min] <- sprintf(glue("%.{d + 1}f"), p[p > min])
new[p <= min] <- sprintf(fmt, p[p <= min])
new
}
source(here::here("analysis/makeTidyGraph.R"))
source(here::here("analysis/plotTidyGraph.R"))config <- here::here("config/config.yml") %>%
read_yaml()
suffix <- paste0(config$tag)
sp <- config$ref$species %>%
str_replace("(^[a-z])[a-z]*_([a-z]+)", "\\1\\2") %>%
str_to_title()topTable <- here::here("output/MFM-223_RNASeq.tsv") %>%
read_tsv()
de <- dplyr::filter(topTable, DE)$gene_id
up <- dplyr::filter(topTable, DE, logFC > 0)$gene_id
down <- dplyr::filter(topTable, DE, logFC < 0)$gene_id
dge <- here::here("output/dge.rds") %>%
read_rds()Setup
Annotations
minPath <- 5
goSummaries <- url("https://uofabioinformaticshub.github.io/summaries2GO/data/goSummaries.RDS") %>%
readRDS() %>%
mutate(exclude = shortest_path < minPath & !terminal_node)msigDB <- msigdbr(species = "Homo sapiens") %>%
dplyr::filter(
gs_cat %in% c("H", "C5") |
gs_subcat %in% c("CP:KEGG", "CP:WIKIPATHWAYS", "TFT:GTRD", "TFT:TFT_Legacy"),
gs_subcat != "HPO"
) %>%
inner_join(
dge$genes %>%
unchop(entrezid) %>%
dplyr::select(
entrez_gene = entrezid,
gene_id
)
) %>%
mutate(
exclude = gs_exact_source %in% dplyr::filter(goSummaries, exclude)$id
)
nSets <- dplyr::filter(msigDB, !exclude)$gs_id %>%
unique() %>%
length()pathByGene <- msigDB %>%
dplyr::filter(gs_cat != "C3", !exclude) %>%
split(.$gene_id) %>%
lapply(pull, "gs_name")
tfByGene <- msigDB %>%
dplyr::filter(gs_cat == "C3", !exclude) %>%
split(.$gene_id) %>%
lapply(pull, "gs_name")genesByPath <- msigDB %>%
dplyr::filter(gs_cat != "C3", !exclude) %>%
split(.$gs_name) %>%
lapply(pull, "gene_id")
genesByTF <- msigDB %>%
dplyr::filter(gs_cat == "C3", !exclude) %>%
split(.$gs_name) %>%
lapply(pull, "gene_id")Gene-set collections were imported from MSigDB version 7.2.1. For gene-sets derived from GO terms, those with fewer than 5 steps back to each ontology root node were excluded as these were likely to be less informative than those at lower levels of the ontology. However, all terms considered as terminal nodes (i.e. with no children) were additionally retained. Information regarding the shortest path back to each ontology root was obtained from https://uofabioinformaticshub.github.io/summaries2GO/MakeSummaries.
The gene-sets belonging to Category C3 were associated with transcriptional regulation, whilst the remaining gene-sets were more focussed on processes and pathways. Two analyses are performed below, following this distinction.
msigDB %>%
distinct(gs_id, .keep_all = TRUE) %>%
mutate(
gs_cat = as.factor(gs_cat) %>% relevel("H"),
gs_subcat = case_when(
gs_cat == "H" ~ "HALLMARK",
TRUE ~ str_replace(gs_subcat, ":", "\\\\:")
)
) %>%
group_by(gs_cat, gs_subcat, exclude) %>%
tally() %>%
ungroup() %>%
pivot_wider(
names_from = exclude,
values_from = n
) %>%
bind_rows(
tibble(
gs_cat = "Total",
gs_subcat = NA,
`FALSE` = sum(.$`FALSE`),
`TRUE` = sum(.$`TRUE`, na.rm = TRUE)
)
) %>%
dplyr::rename(
Category = gs_cat,
Collection = gs_subcat,
`Retained Gene-Sets` = `FALSE`,
`Discarded Gene Sets`= `TRUE`
) %>%
pander(
justify = "llrr",
emphasize.strong.rows = nrow(.),
caption = glue(
"*Summary of gene-sets and collections used in this analysis.
For a GO term to be retained, it was required to be a terminal node,
or have a shortest path back to the root node of {minPath} or more steps.*"
)
)| Category | Collection | Retained Gene-Sets | Discarded Gene Sets |
|---|---|---|---|
| H | HALLMARK | 50 | |
| C2 | CP:KEGG | 186 | |
| C2 | CP:WIKIPATHWAYS | 583 | |
| C3 | TFT:GTRD | 347 | |
| C3 | TFT:TFT_Legacy | 610 | |
| C5 | GO:BP | 4,160 | 3,362 |
| C5 | GO:CC | 487 | 508 |
| C5 | GO:MF | 1,127 | 530 |
| Total | 7,550 | 4,400 |
Assessment of Sampling Bias for DE Genes
pwf <- list(
length = topTable %>%
arrange(gene_id) %>%
with(
nullp(
DEgenes = structure(DE, names = gene_id),
bias.data = ave_tx_len,
plot.fit = FALSE
)
),
gc = topTable %>%
arrange(gene_id) %>%
with(
nullp(
DEgenes = structure(DE, names = gene_id),
bias.data = gc_content,
plot.fit = FALSE
)
)
)par(mfrow = c(1, 2))
plotPWF(pwf$length, xlab = "Gene Length", ylim = c(0, 0.13), log = "x")
plotPWF(pwf$gc, xlab = "GC content", ylim = c(0, 0.13))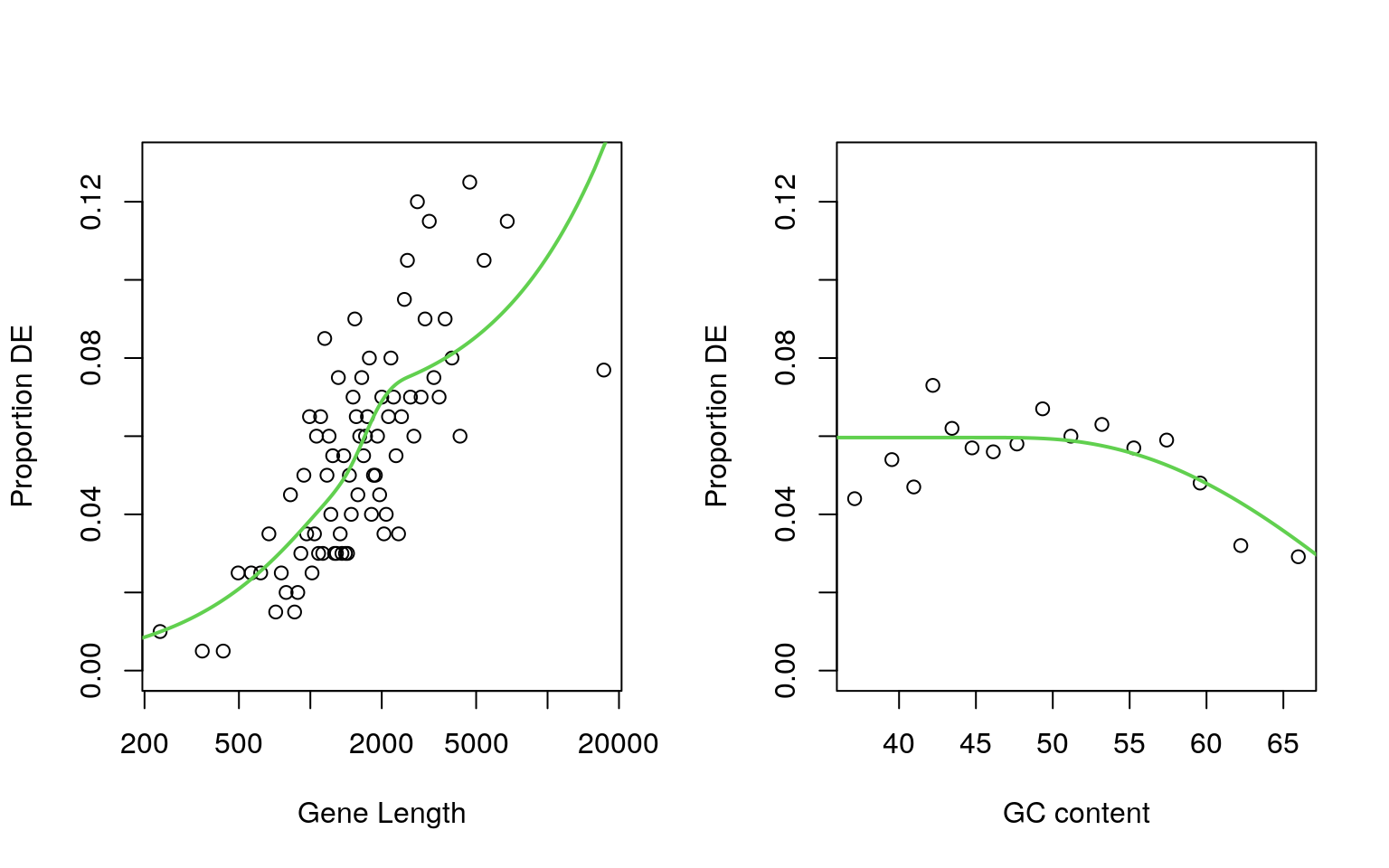
Comparison of gene length and GC content on the probability of a gene being considered as DE
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
par(mfrow = c(1, 1))As some bias has been previously identified in this dataset, a probability weight function for consideration of a gene as DE was estimated using the standard goseq workflow. Both GC content and gene length were investigated as potential sources of bias with gene length demonstrating the largest influence. This was subsequently included as an offset for sampling bias in all enrichment analyses looking within the set of DE genes.
For enrichment analysis of up and down-regulated genes separately, probability weight functions were also generated for both subsets of DE genes
upPwf <- topTable %>%
arrange(gene_id) %>%
with(
nullp(
DEgenes = structure(DE & logFC > 0, names = gene_id),
bias.data = ave_tx_len,
plot.fit = FALSE
)
)
downPwf <- topTable %>%
arrange(gene_id) %>%
with(
nullp(
DEgenes = structure(DE & logFC < 0, names = gene_id),
bias.data = ave_tx_len,
plot.fit = FALSE
)
)
par(mfrow = c(1, 2))
plotPWF(upPwf, main = "Up-regulated genes", log = "x", ylim = c(0, 0.1))
plotPWF(downPwf, main = "Down-regulated genes", log = "x", ylim = c(0, 0.1))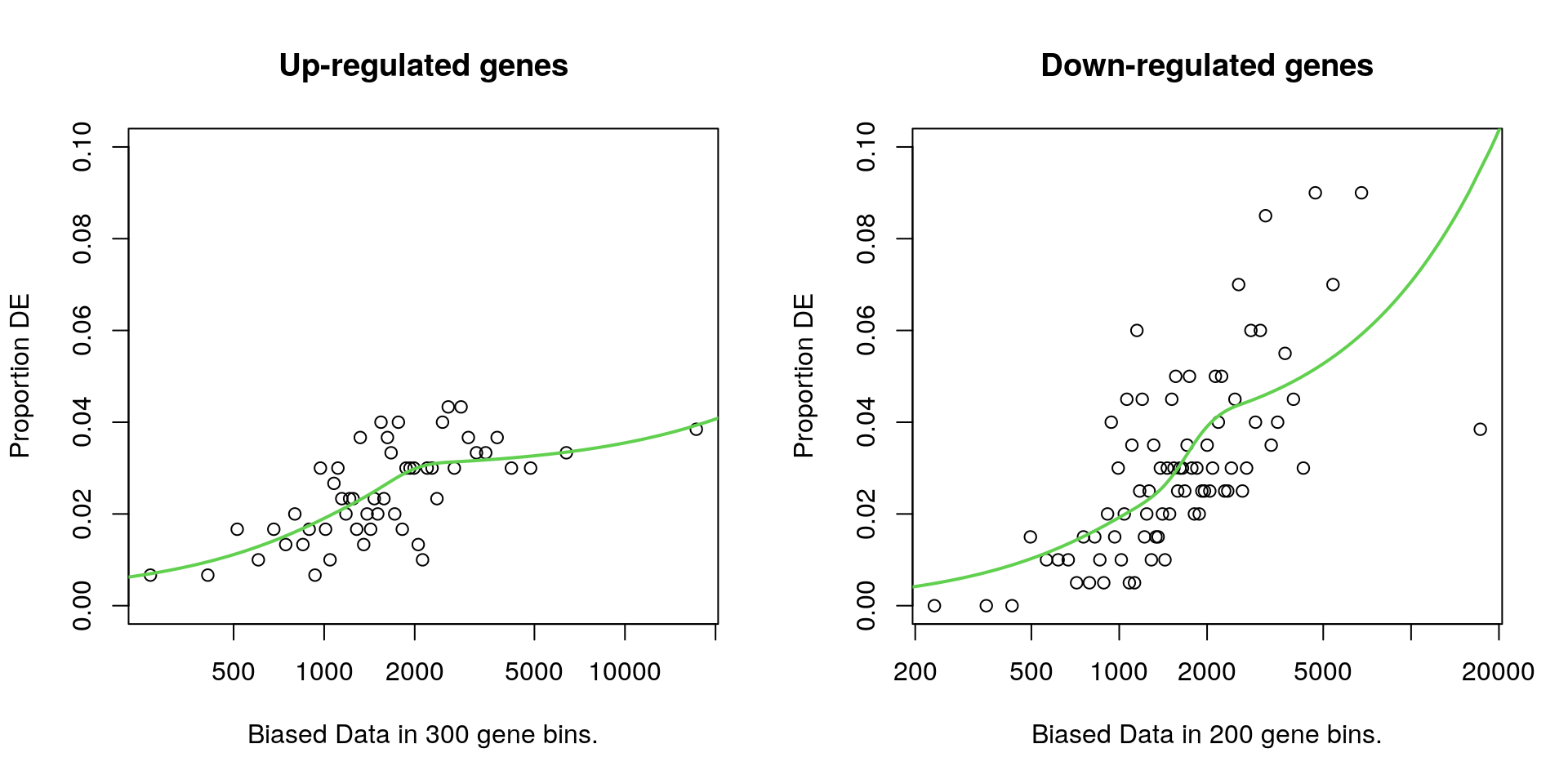
The influence of gene length on the probability of being considered DE, for both up and down-regulated genes
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
par(mfrow = c(1, 1))Pathway and Process Focussed Analysis
All DE Genes
pathGoseqRes <- goseq(pwf$length, gene2cat = pathByGene) %>%
as_tibble() %>%
dplyr::mutate(
Expected = round(sum(topTable$DE) * numInCat / nrow(topTable), 0),
FDR = p.adjust(over_represented_pvalue, "BH")
) %>%
dplyr::select(
Category = category,
`Number DE` = numDEInCat,
Expected,
`Gene Set Size` = numInCat,
`Enrichment p` = over_represented_pvalue,
FDR
)
alpha <- 0.01Using an FDR threshold of \(\alpha =\) 0.01, 9 pathway & process-related gene sets were considered as enriched within the set of 856 previously-defined DE genes.
alpha <- 0.01
sigPath <- genesByPath %>%
.[dplyr::filter(pathGoseqRes, FDR < alpha)$Category] %>%
lapply(intersect, de) %>%
lapply(function(x){dge$genes[x,]$gene_name}) %>%
setNames(
names(.) %>%
str_replace_all("(HALLMARK|GO|KEGG|WP)_(.+)", "\\2 (\\1)") %>%
str_replace_all("_", " ") %>%
str_wrap(16)
)
pathGraph <- makeTidyGraph(sigPath, topTable)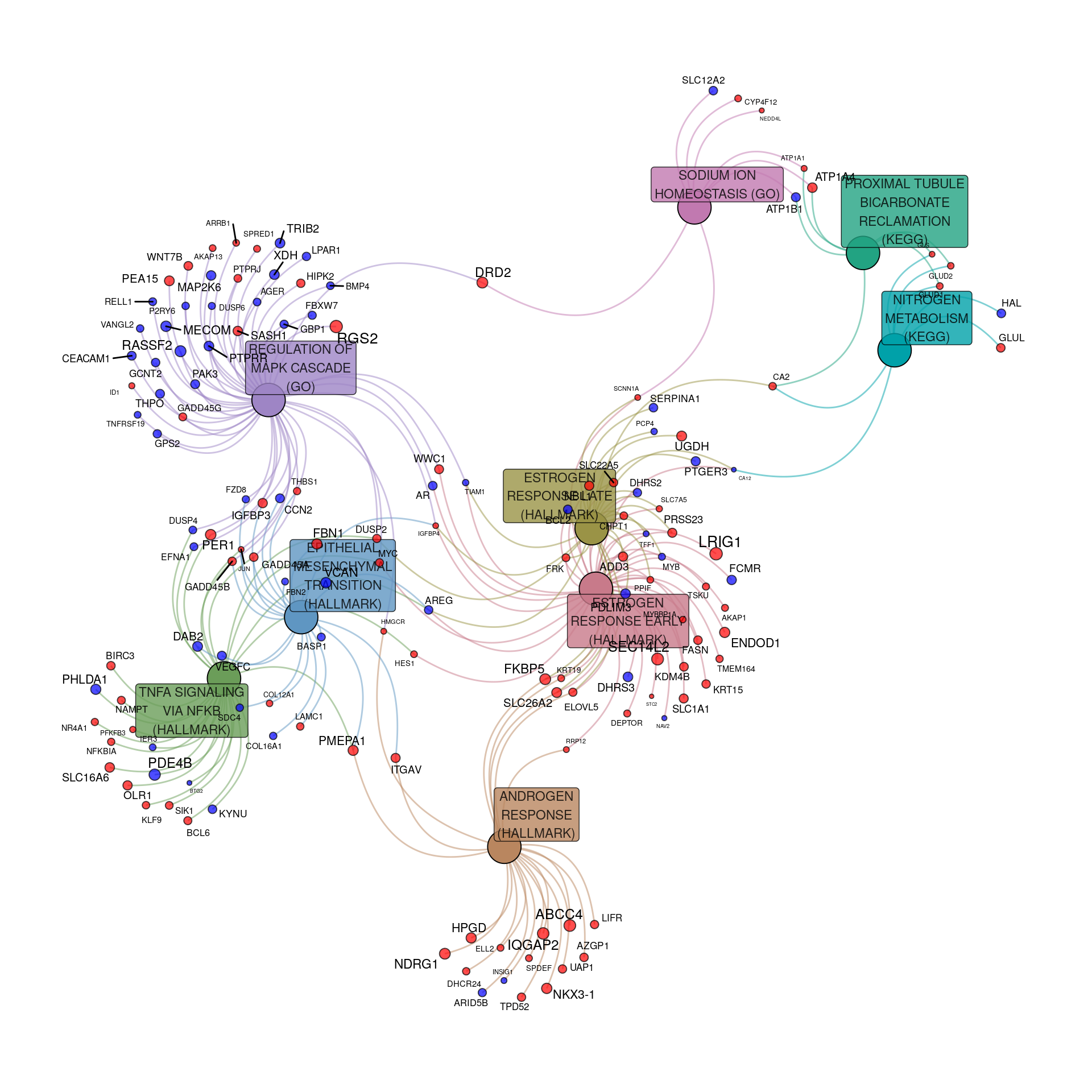
Pathway & process-related gene-sets enriched within the DE genes to an FDR of 0.01. Up-regulated genes are shown in red, with down-regulated in blue. For genes, node and label size are proportional to the extent of the estimated fold-change.
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
The above visualisation clearly showed:
- The Androgen Response was primarily up-regulated
- The Early and Late Estrogen Responses were both observed, and largely overlapped
- Regulation of the MAPK Cascade, TNF\(\alpha\) signalling via NF\(\kappa\beta\) and the EMT transition were highly connected
- Three connected metabolic processes were also well connected
msigDB %>%
dplyr::filter(
gs_name %in% dplyr::filter(pathGoseqRes, FDR < alpha)$Category
) %>%
mutate(
direction = case_when(
gene_id %in% up ~ "Up",
gene_id %in% down ~ "Down",
TRUE ~ "Unchanged"
) %>%
factor(levels = c("Unchanged", "Down", "Up"))
) %>%
group_by(gs_name, direction) %>%
tally() %>%
ungroup() %>%
arrange(desc(n)) %>%
mutate(gs_name = fct_inorder(gs_name)) %>%
ggplot(
aes(gs_name, n, fill = direction)
) +
geom_col() +
coord_flip() +
labs(
x = "Gene Set",
y = "Number of Genes",
fill = "Direction"
) +
scale_y_continuous(expand = expansion(c(0, 0.05))) +
scale_fill_manual(
values = c("grey80", "blue", "red")
) +
theme(
legend.position = c(1, 1)*0.99,
legend.justification = c(1, 1),
panel.grid.major.y = element_blank()
)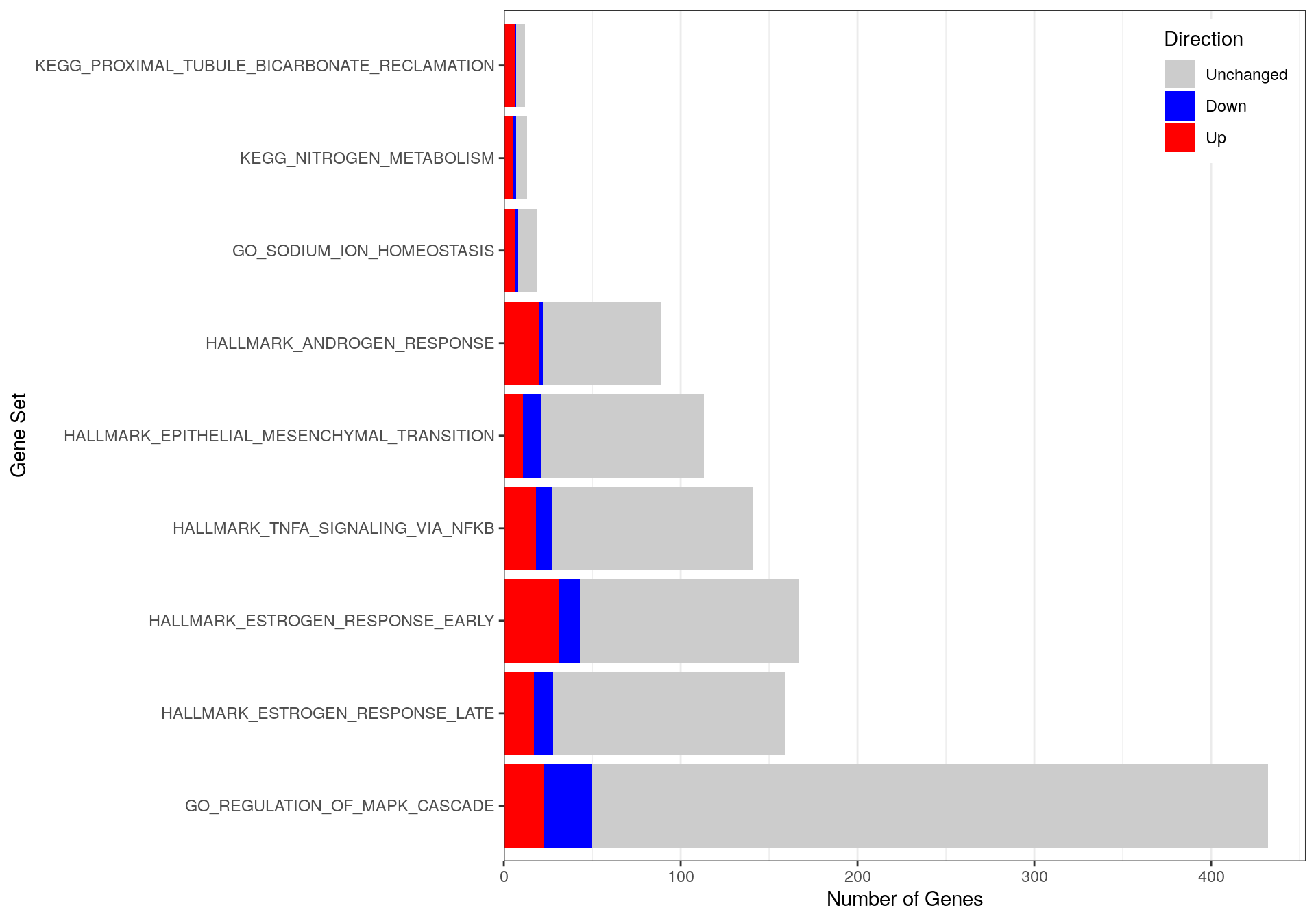
Distribution of up & down-regulated genes within each enriched pathway & process-related gene-set.
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
Up-Regulated Genes
upPathGoseqRes <- goseq(upPwf, gene2cat = pathByGene) %>%
as_tibble() %>%
dplyr::mutate(
Expected = round(length(up) * numInCat / nrow(topTable), 0),
FDR = p.adjust(over_represented_pvalue, "BH")
) %>%
dplyr::select(
Category = category,
`Number DE` = numDEInCat,
Expected,
`Gene Set Size` = numInCat,
`Enrichment p` = over_represented_pvalue,
FDR
)
alpha <- 0.05Given the reduced power of the smaller gene-sets used when investigating up & down-regulated genes separately, an FDR of 0.05 was chosen for this section. Using this FDR threshold of \(\alpha =\) 0.05, 33 pathway & process-related gene sets were considered as enriched within the set of 384 up-regulated genes.
alpha <- 0.05
minSize <- 5
sigPath <- genesByPath %>%
.[dplyr::filter(upPathGoseqRes, FDR < alpha)$Category] %>%
lapply(intersect, up) %>%
lapply(function(x){dge$genes[x,]$gene_name}) %>%
setNames(
names(.) %>%
str_replace_all("(HALLMARK|GO|KEGG|WP)_(.+)", "\\2 (\\1)") %>%
str_replace_all("_", " ") %>%
str_wrap(16)
) %>%
.[vapply(., length, integer(1)) >= minSize]
pathGraph <- makeTidyGraph(sigPath, topTable)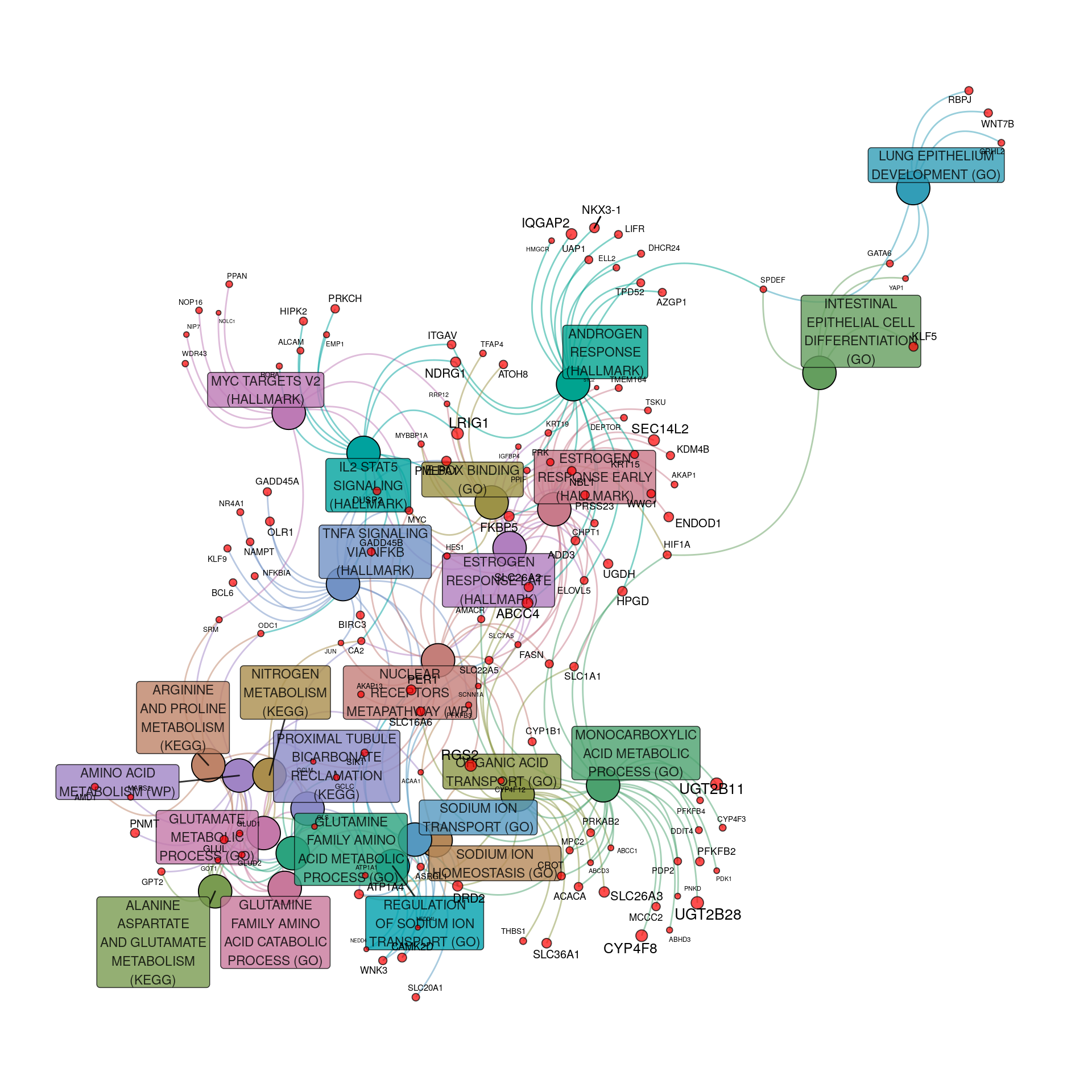
Pathway & process-related gene-sets enriched within the set of up-regulated genes to an FDR of 0.05. For genes, node and label size are proportional to the extent of the estimated fold-change. Gene-sets are only included if they contain at least 5 up-regulated genes
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
Results for process & pathway analysis in up-regulated genes once again showed
- The large set of interconnected metabolic processes
- A cluster of related pathways including ER and AR responses, along with IL2 and TNF\(\alpha\) signalling
- The MYC Targets pathway was somewhat less connected
- Epithelial Development pathways were also less connected
Down-Regulated Genes
downPathGoseqRes <- goseq(downPwf, gene2cat = pathByGene) %>%
as_tibble() %>%
dplyr::mutate(
Expected = round(length(down) * numInCat / nrow(topTable), 0),
FDR = p.adjust(over_represented_pvalue, "BH")
) %>%
dplyr::select(
Category = category,
`Number DE` = numDEInCat,
Expected,
`Gene Set Size` = numInCat,
`Enrichment p` = over_represented_pvalue,
FDR
)
alpha <- 0.05Using an FDR threshold of \(\alpha =\) 0.05, 0 pathway & process-related gene sets were considered as enriched within the set of 472 down-regulated genes.
Combined Up And Down-Regulated Results
bind_rows(
upPathGoseqRes %>% mutate(Direction = "Up"),
downPathGoseqRes %>% mutate(Direction = "Down")
) %>%
group_by(Category) %>%
dplyr::filter(min(FDR) < alpha) %>%
arrange(mean(1/ `Enrichment p`, na.rm = TRUE)) %>%
ungroup() %>%
mutate(
Category = case_when(
Category %in% dplyr::filter(pathGoseqRes, FDR<0.05)$Category ~ paste0("*", Category),
TRUE ~ Category
),
Category = fct_inorder(Category)
) %>%
ggplot(aes(Category, -log10(`Enrichment p`), fill = Direction)) +
geom_col(position = "dodge") +
geom_hline(yintercept = 3, linetype = 2) +
coord_flip() +
labs(y = expression(paste(log[10], "p"))) +
scale_y_continuous(expand = expansion(c(0, 0.05))) +
scale_fill_manual(
values = c("#0000FFB3", "#FF0000B3")
) +
theme(
legend.position = c(1, 1)*0.99,
legend.justification = c(1, 1)
)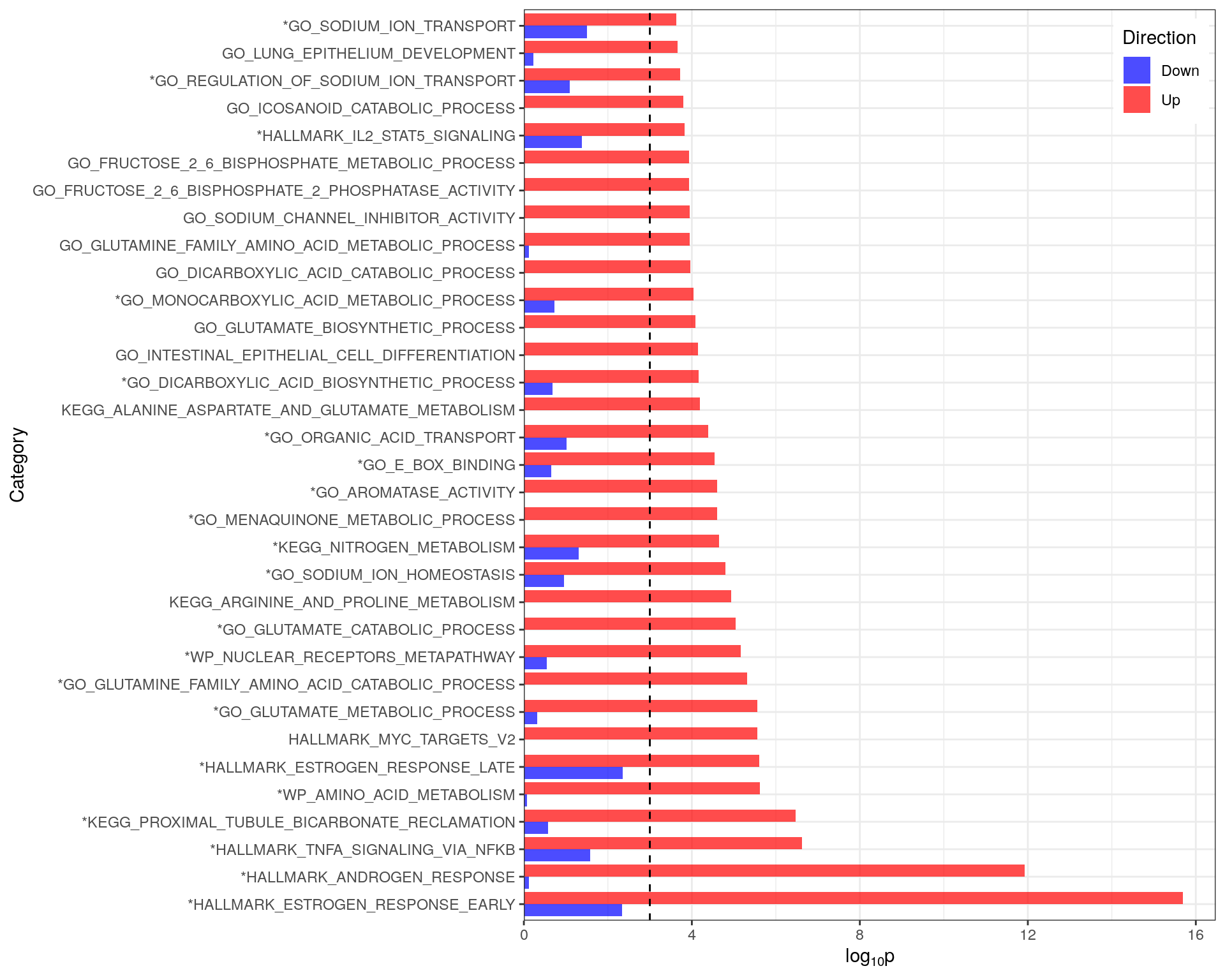
Comparison of p-values for directional enrichment showing that all enrichment results had a clear directional bias. Only pathways with an FDR-adjusted p-value < 0.05 are shown. The vertical black line indicates a raw p-value of 0.001 as results which fail to acheive this value are usually non-significant. Gene-sets considered as significant in the combined (i.e. non-directional) analysis are marked with an asterisk
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
A comparison of the p-values obtained from the individual directional analysis revealed that all pathways showed a clear directional bias, with each pathway only enriched in one direction.
Regulation-Focussed Analysis
All DE Genes
tfGoseqRes <- goseq(pwf$length, gene2cat = tfByGene) %>%
as_tibble() %>%
dplyr::mutate(
Expected = round(sum(topTable$DE) * numInCat / nrow(topTable), 0),
FDR = p.adjust(over_represented_pvalue, "BH")
) %>%
dplyr::select(
Category = category,
`Number DE` = numDEInCat,
Expected,
`Gene Set Size` = numInCat,
`Enrichment p` = over_represented_pvalue,
FDR
)
alpha <- 0.01Using an FDR threshold of \(\alpha =\) 0.01, 18 transcriptional regulation gene sets were considered as enriched within the set of 856 previously defined DE genes.
alpha <- 0.01
sigTF <- genesByTF %>%
.[dplyr::filter(tfGoseqRes, FDR < alpha)$Category] %>%
# .[dplyr::slice(tfGoseqRes, 1:15)$Category] %>%
lapply(intersect, de) %>%
lapply(function(x){dge$genes[x,]$gene_name}) %>%
setNames(
names(.) %>%
str_replace_all("(HALLMARK|GO|KEGG|WP)_(.+)", "\\2 (\\1)") %>%
str_replace_all("_", " ") %>%
str_wrap(16)
)
tfGraph <- makeTidyGraph(sigTF, topTable)
Transcriptional regulatory gene-sets enriched within the DE genes to an FDR of 0.01. Up-regulated genes are shown in red, with down-regulated in blue. For genes, node and label size are proportional to the extent of the estimated fold-change.
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
Whilst the above network plot initially appears uninformative, it can be seen that genes under regulatory control by GREB1, NFAT and AP1 appear relatively distinct
This is supported by the correlation plot highlighting the trend of co-occurrence for all significant TF motifs. LEF1 and TCF4 appear to co-occur a noticeable amount, with CEBP also showing some association with CREL, MEF2 and NKX6.
tfGoseqRes %>%
dplyr::filter(FDR < alpha) %>%
dplyr::select(gs_name = Category) %>%
left_join(msigDB) %>%
dplyr::filter(gene_id %in% de) %>%
dplyr::mutate(is_in = TRUE) %>%
pivot_wider(
id_cols = c("gs_name", "gene_symbol", "gene_id"),
names_from = gs_name,
values_from = is_in,
values_fill = FALSE
) %>%
dplyr::select(any_of(msigDB$gs_name)) %>%
cor() %>%
corrplot(
type = "upper",
diag = FALSE,
addCoef.col = rgb(0, 0, 0, 0.6),
addCoefasPercent = TRUE,
order = "hclust",
addshade = "all",
col = colorRampPalette(c("blue", "white", "red"))(100),
tl.cex = 0.7,
number.cex = 0.7
)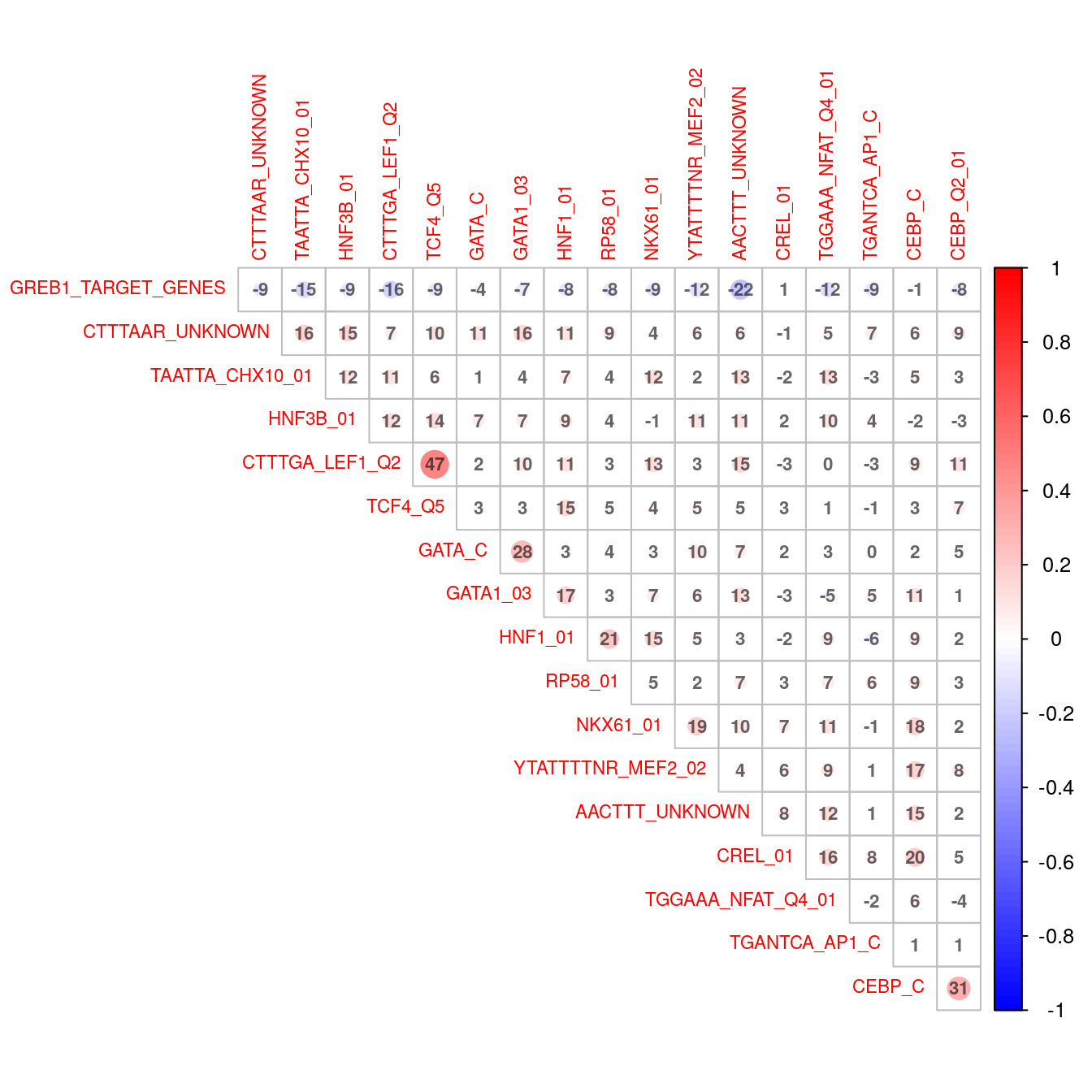
Correlation plot indicating co-occurence between all enriched transcription factor motifs (FDR < 0.01).
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
Up-Regulated Genes
upTfGoseqRes <- goseq(upPwf, gene2cat = tfByGene) %>%
as_tibble() %>%
dplyr::mutate(
Expected = round(length(up)* numInCat / nrow(topTable), 0),
FDR = p.adjust(over_represented_pvalue, "BH")
) %>%
dplyr::select(
Category = category,
`Number DE` = numDEInCat,
Expected,
`Gene Set Size` = numInCat,
`Enrichment p` = over_represented_pvalue,
FDR
)
alpha <- 0.05Given the reduced power of the smaller gene-sets used when investigating up & down-regulated genes separately, an FDR of 0.05 was again chosen for this section. Using this FDR threshold of \(\alpha =\) 0.05, 76 transcriptional regulation gene sets were considered as enriched within the set of 384 previously defined up-regulated genes.
sigTF <- genesByTF %>%
.[dplyr::filter(upTfGoseqRes, FDR < alpha)$Category] %>%
lapply(intersect, up) %>%
lapply(function(x){dge$genes[x,]$gene_name}) %>%
setNames(
names(.) %>%
str_replace_all("(HALLMARK|GO|KEGG|WP)_(.+)", "\\2 (\\1)") %>%
str_replace_all("_", " ") %>%
str_wrap(16)
)
tfGraph <- makeTidyGraph(sigTF, topTable)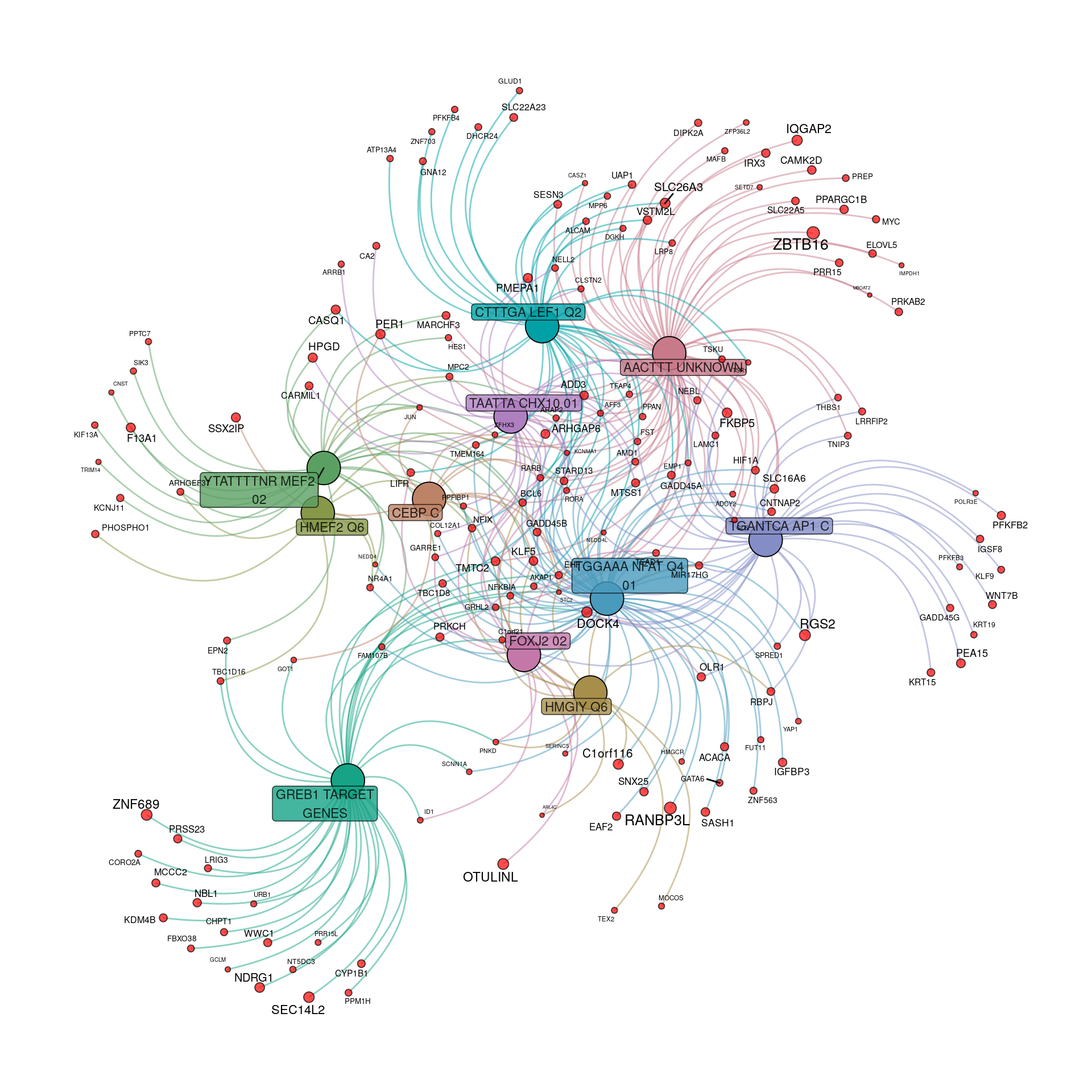
Transcriptional regulatory gene-sets enriched within the 384 up-regulated genes to an FDR of 0.05. For genes, node and label size are proportional to the extent of the estimated fold-change.
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
The distinct patterns of regulation by GREB1 and LEF1 were noticeable in both the above network plot and the correlations.
upTfGoseqRes %>%
dplyr::filter(FDR < alpha) %>%
dplyr::select(gs_name = Category) %>%
left_join(msigDB) %>%
dplyr::filter(gene_id %in% up) %>%
dplyr::mutate(is_in = TRUE) %>%
pivot_wider(
id_cols = c("gs_name", "gene_symbol", "gene_id"),
names_from = gs_name,
values_from = is_in,
values_fill = FALSE
) %>%
dplyr::select(any_of(msigDB$gs_name)) %>%
cor() %>%
corrplot(
type = "upper",
diag = FALSE,
addCoef.col = rgb(0, 0, 0, 0.6),
addCoefasPercent = TRUE,
order = "hclust",
addshade = "all",
col = colorRampPalette(c("blue", "white", "red"))(100),
tl.cex = 0.7,
number.cex = 0.7
)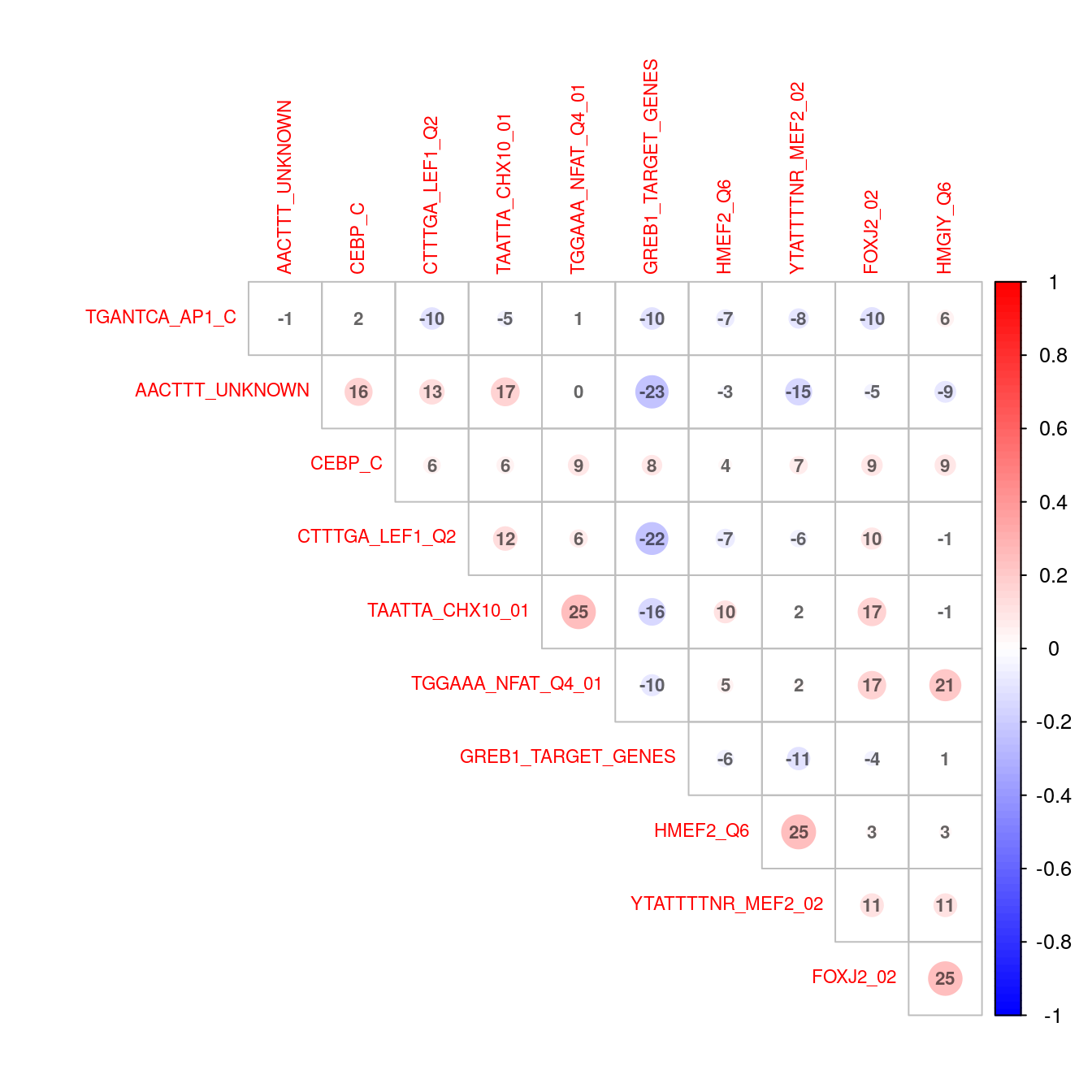
Correlation plot indicating co-occurence between all enriched transcription factor motifs (FDR < 0.05) in up-regulated genes.
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
Down-Regulated Genes
downTfGoseqRes <- goseq(downPwf, gene2cat = tfByGene) %>%
as_tibble() %>%
dplyr::mutate(
Expected = round(length(down)* numInCat / nrow(topTable), 0),
FDR = p.adjust(over_represented_pvalue, "BH")
) %>%
dplyr::select(
Category = category,
`Number DE` = numDEInCat,
Expected,
`Gene Set Size` = numInCat,
`Enrichment p` = over_represented_pvalue,
FDR
)
alpha <- 0.05Given the reduced power of the smaller gene-sets used when investigating up & down-regulated genes separately, an FDR of 0.05 was again chosen for this section. Using this FDR threshold of \(\alpha =\) 0.05, 76 transcriptional regulation gene sets were considered as enriched within the set of 472 previously defined down-regulated genes.
alpha <- 0.05
minSize <- 5
sigTF <- genesByTF %>%
.[dplyr::filter(downTfGoseqRes, FDR < alpha)$Category] %>%
lapply(intersect, down) %>%
lapply(function(x){dge$genes[x,]$gene_name}) %>%
setNames(
names(.) %>%
str_replace_all("(HALLMARK|GO|KEGG|WP)_(.+)", "\\2 (\\1)") %>%
str_replace_all("_", " ") %>%
str_wrap(16)
) %>%
.[vapply(., length, integer(1)) >= minSize]
tfGraph <- makeTidyGraph(sigTF, topTable)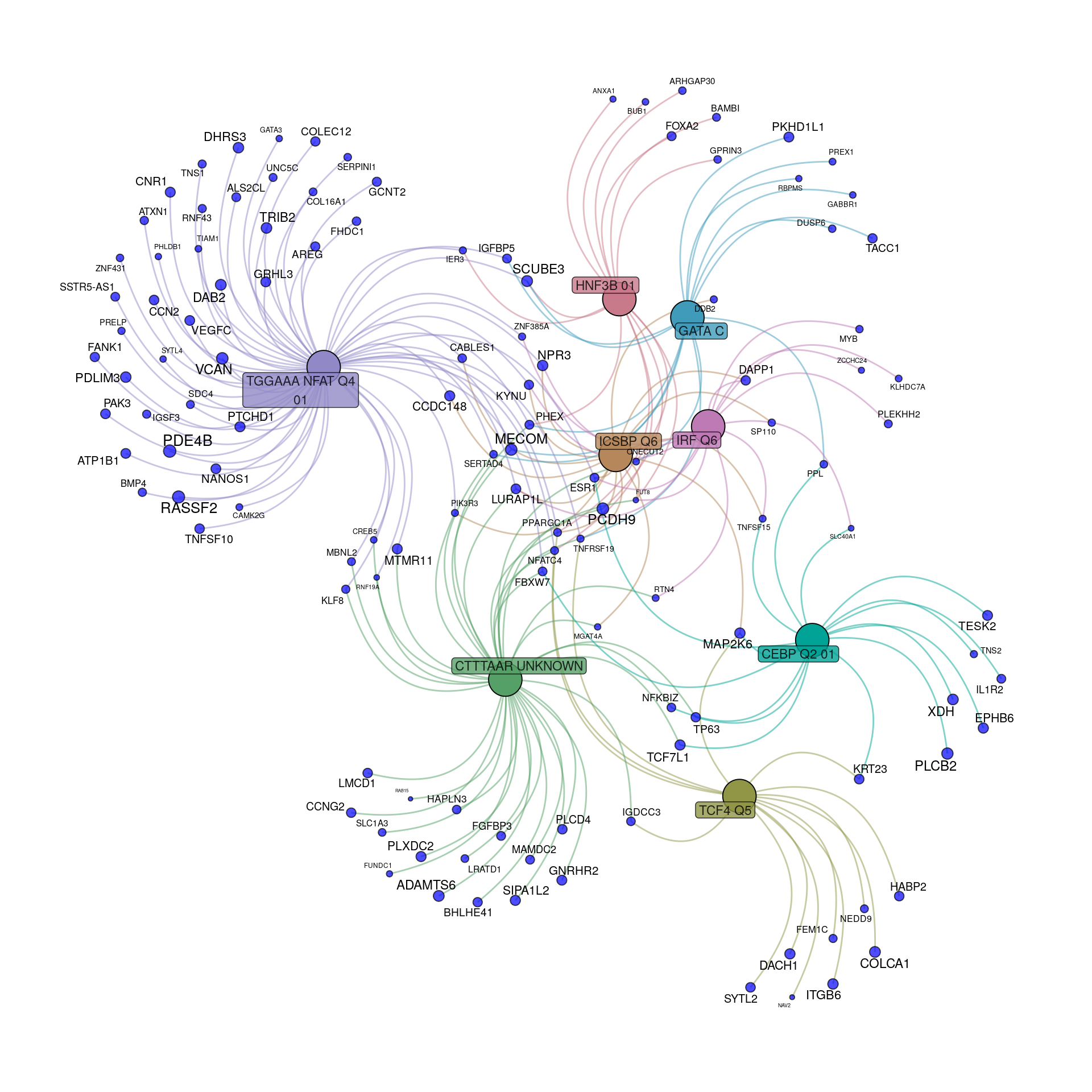
Transcriptional regulatory gene-sets enriched within the 472 down-regulated genes to an FDR of 0.05. For genes, node and label size are proportional to the extent of the estimated fold-change.
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
- CEBP_Q2 and NFATQ4 appear to capture distinct sets of genes
- ICSBP_Q6 and IRF_Q6 appear to capture more common sets of genes
downTfGoseqRes %>%
dplyr::filter(FDR < alpha) %>%
dplyr::select(gs_name = Category) %>%
left_join(msigDB) %>%
dplyr::filter(gene_id %in% down) %>%
dplyr::mutate(is_in = TRUE) %>%
pivot_wider(
id_cols = c("gs_name", "gene_symbol", "gene_id"),
names_from = gs_name,
values_from = is_in,
values_fill = FALSE
) %>%
dplyr::select(any_of(msigDB$gs_name)) %>%
cor() %>%
corrplot(
type = "upper",
diag = FALSE,
addCoef.col = rgb(0, 0, 0, 0.6),
addCoefasPercent = TRUE,
order = "hclust",
addshade = "all",
col = colorRampPalette(c("blue", "white", "red"))(100),
tl.cex = 0.7,
number.cex = 0.7
)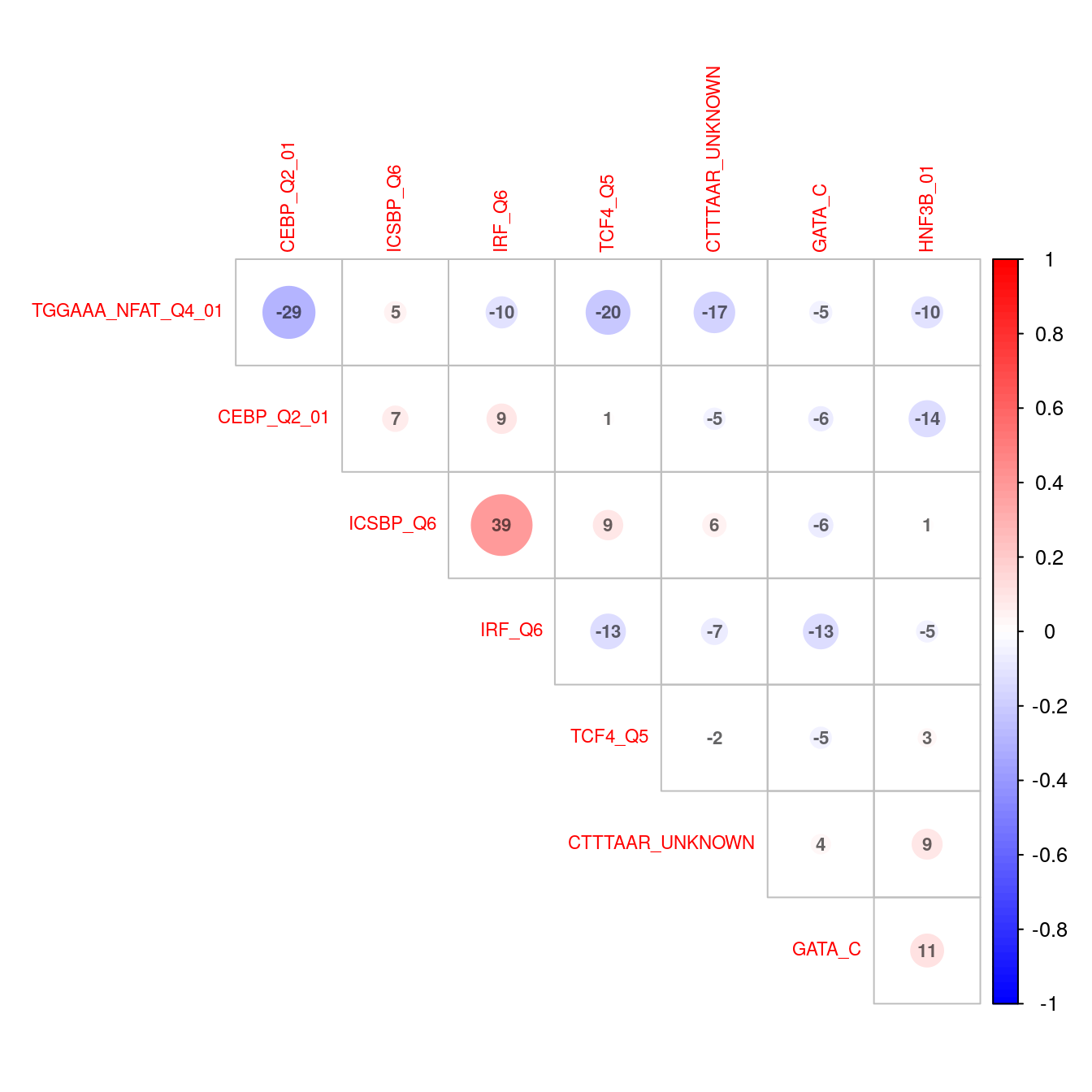
Correlation plot indicating co-occurence between all enriched transcription factor motifs (FDR < 0.05) in down-regulated genes.
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
Combined Up And Down-Regulated Results
alpha <- 0.05bind_rows(
upTfGoseqRes %>% mutate(Direction = "Up"),
downTfGoseqRes %>% mutate(Direction = "Down")
) %>%
group_by(Category) %>%
dplyr::filter(min(FDR) < alpha & min(`Enrichment p`) < 0.001) %>%
arrange(mean(1/ `Enrichment p`, na.rm = TRUE)) %>%
ungroup() %>%
mutate(Category = fct_inorder(Category)) %>%
ggplot(aes(Category, -log10(`Enrichment p`), fill = Direction)) +
geom_col(position = "dodge") +
geom_hline(yintercept = 3, linetype = 2) +
coord_flip() +
labs(y = expression(paste(log[10], "p"))) +
scale_y_continuous(expand = expansion(c(0, 0.05))) +
scale_fill_manual(
values = c("#0000FFB3", "#FF0000B3")
) +
theme(
legend.position = c(1, 1)*0.99,
legend.justification = c(1, 1)
)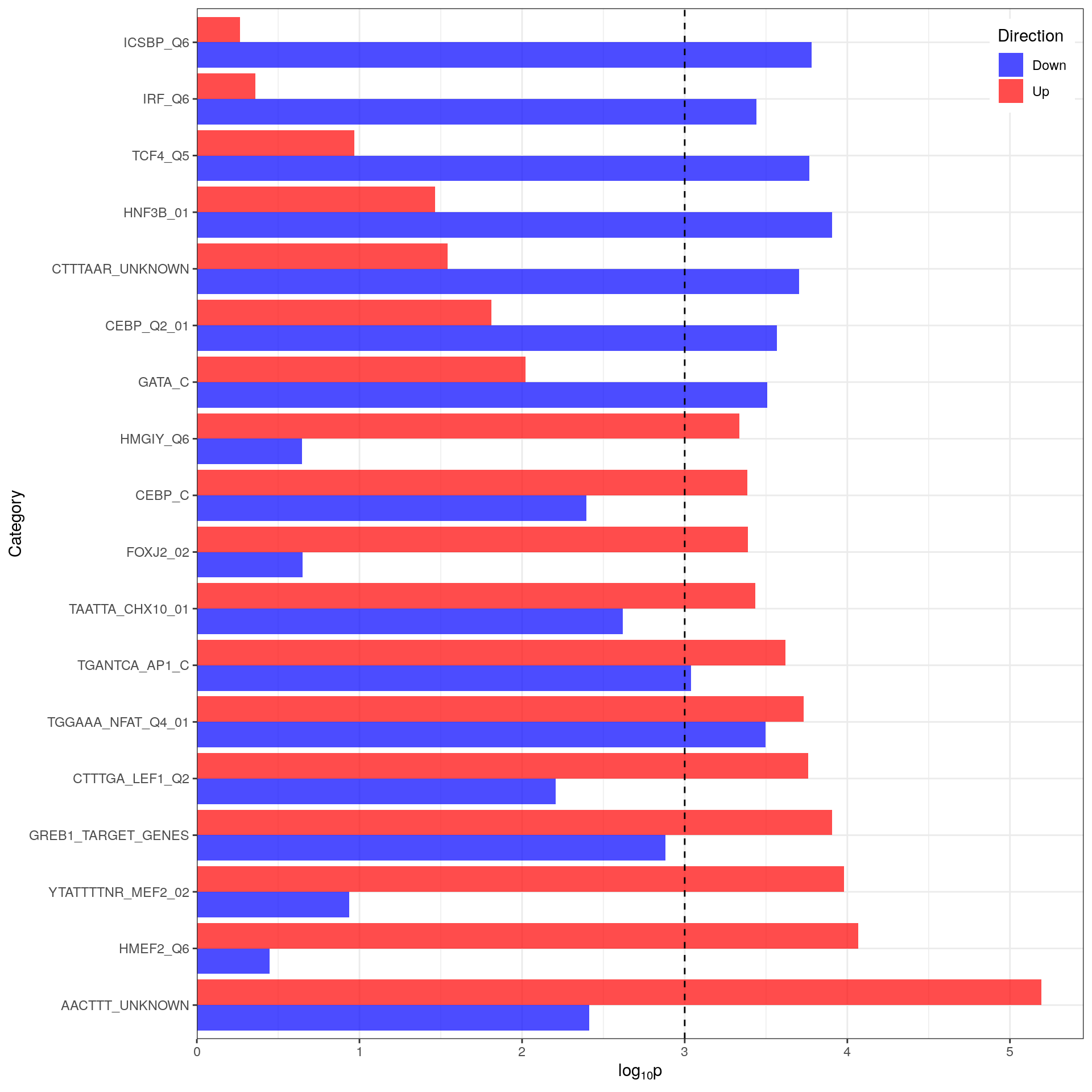
Comparison of p-values for directional enrichment. Only gene-sets with an FDR-adjusted p-value < 0.05 are shown. The vertical black line indicates a raw p-value of 0.001 as results which fail to acheive this value are usually non-significant. All gene-sets were considered significant in the non-directional analysis.
| Version | Author | Date |
|---|---|---|
| bcdc500 | Steve Ped | 2020-10-15 |
A comparison of the p-values obtained from the individual directional analysis revealed:
- Both AP1 and NFAT regulated genes appear to have no directional bias
- GREB1, LEF1 and CHX10 regulated genes appear slightly biased towards up-regulation
- Most other gene-sets have a bias to either up (e.g. AACTTT_UNKNOWN, HMEF2) or down (e.g. ICSBP, IRF, TCF4) regulation
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.5 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] corrplot_0.84 ggraph_2.0.3 tidygraph_1.2.0
[4] htmltools_0.5.0 reactable_0.2.3 goseq_1.40.0
[7] geneLenDataBase_1.24.0 BiasedUrn_1.07 msigdbr_7.2.1
[10] DT_0.15 ggrepel_0.8.2 magrittr_1.5
[13] cowplot_1.1.0 edgeR_3.30.3 limma_3.44.3
[16] glue_1.4.2 pander_0.6.3 scales_1.1.1
[19] yaml_2.2.1 forcats_0.5.0 stringr_1.4.0
[22] dplyr_1.0.2 purrr_0.3.4 readr_1.4.0
[25] tidyr_1.1.2 tibble_3.0.3 ggplot2_3.3.2
[28] tidyverse_1.3.0 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] readxl_1.3.1 backports_1.1.10
[3] BiocFileCache_1.12.1 plyr_1.8.6
[5] igraph_1.2.6 splines_4.0.3
[7] crosstalk_1.1.0.1 BiocParallel_1.22.0
[9] usethis_1.6.3 GenomeInfoDb_1.24.2
[11] digest_0.6.25 viridis_0.5.1
[13] GO.db_3.11.4 fansi_0.4.1
[15] memoise_1.1.0 remotes_2.2.0
[17] Biostrings_2.56.0 graphlayouts_0.7.0
[19] modelr_0.1.8 matrixStats_0.57.0
[21] askpass_1.1 prettyunits_1.1.1
[23] colorspace_1.4-1 blob_1.2.1
[25] rvest_0.3.6 rappdirs_0.3.1
[27] haven_2.3.1 xfun_0.18
[29] callr_3.4.4 crayon_1.3.4
[31] RCurl_1.98-1.2 jsonlite_1.7.1
[33] polyclip_1.10-0 gtable_0.3.0
[35] zlibbioc_1.34.0 XVector_0.28.0
[37] DelayedArray_0.14.1 pkgbuild_1.1.0
[39] BiocGenerics_0.34.0 DBI_1.1.0
[41] Rcpp_1.0.5 viridisLite_0.3.0
[43] progress_1.2.2 bit_4.0.4
[45] stats4_4.0.3 htmlwidgets_1.5.2
[47] httr_1.4.2 ellipsis_0.3.1
[49] pkgconfig_2.0.3 XML_3.99-0.5
[51] farver_2.0.3 dbplyr_1.4.4
[53] locfit_1.5-9.4 here_0.1
[55] labeling_0.3 tidyselect_1.1.0
[57] rlang_0.4.7 later_1.1.0.1
[59] AnnotationDbi_1.50.3 reactR_0.4.3
[61] munsell_0.5.0 cellranger_1.1.0
[63] tools_4.0.3 cli_2.0.2
[65] generics_0.0.2 RSQLite_2.2.1
[67] devtools_2.3.2 broom_0.7.1
[69] evaluate_0.14 processx_3.4.4
[71] knitr_1.30 bit64_4.0.5
[73] fs_1.5.0 nlme_3.1-149
[75] whisker_0.4 xml2_1.3.2
[77] biomaRt_2.44.1 compiler_4.0.3
[79] rstudioapi_0.11 curl_4.3
[81] testthat_2.3.2 reprex_0.3.0
[83] tweenr_1.0.1 stringi_1.5.3
[85] highr_0.8 ps_1.3.4
[87] GenomicFeatures_1.40.1 desc_1.2.0
[89] lattice_0.20-41 Matrix_1.2-18
[91] vctrs_0.3.4 pillar_1.4.6
[93] lifecycle_0.2.0 bitops_1.0-6
[95] httpuv_1.5.4 rtracklayer_1.48.0
[97] GenomicRanges_1.40.0 R6_2.4.1
[99] promises_1.1.1 gridExtra_2.3
[101] IRanges_2.22.2 sessioninfo_1.1.1
[103] MASS_7.3-53 assertthat_0.2.1
[105] pkgload_1.1.0 SummarizedExperiment_1.18.2
[107] openssl_1.4.3 rprojroot_1.3-2
[109] withr_2.3.0 GenomicAlignments_1.24.0
[111] Rsamtools_2.4.0 S4Vectors_0.26.1
[113] GenomeInfoDbData_1.2.3 mgcv_1.8-33
[115] parallel_4.0.3 hms_0.5.3
[117] grid_4.0.3 rmarkdown_2.4
[119] git2r_0.27.1 ggforce_0.3.2
[121] Biobase_2.48.0 lubridate_1.7.9